- Introduction
- Materialien und Methoden
- Studienproben
- Genomische DNA- und RNA-Extraktionen
- Bibliotheksvorbereitung und Sequenzierung
- gDNA-Capture mit IDT Xgen® Lockdown®-Sonden und Einzelmolekül-Sequenzierung
- CDNA-Capture mit IDT Xgen® Lockdown®-Sonden und Einzelmolekül-Isoform-Sequenzierung (Iso-Seq)
- gDNA-Analyse
- Kurzvariantenanalyse und Phasing
- Clustering und Bestimmung von Haplotypen für CT-Rich Region
- Isoform-Analyse
- Isoform SNP Calling
- Ergebnisse
- Gezieltes gDNA-Capture identifizierte bekannte und neue Variationen
- Targeted cDNA Capture Identified Novel Start and End Sites
- CDNA in voller Länge ermöglicht Phasing-Informationen auf Isoform-Ebene
- Diskussion
- Datenverfügbarkeit
- Beiträge der Autoren
- Finanzierung
- Erklärung zu Interessenkonflikten
- Danksagungen
- Ergänzendes Material
Introduction
Transkriptionelle und posttranskriptionelle Programme kontrollieren das Niveau der Genexpression und/oder die Produktion mehrerer unterschiedlicher mRNA-Isoformen, und Veränderungen in diesen Mechanismen führen zu einer Dysregulation der Genexpression und zu unterschiedlichen Expressionsprofilen. Eine abweichende transkriptionelle und posttranskriptionelle Genregulation ist in den Geweben des menschlichen Nervensystems häufig anzutreffen und trägt zu phänotypischen Unterschieden innerhalb und zwischen Individuen in Gesundheit und Krankheit bei.
Die Dysregulation der Alpha-Synuclein-Expression wurde mit der Pathogenese von Synucleinopathien in Verbindung gebracht, insbesondere mit der Parkinson-Krankheit (PD) und der Demenz mit Lewy-Körpern (DLB). Während die Rolle der SNCA-Überexpression bei Synucleinopathien, vor allem bei Morbus Parkinson, gut belegt ist, haben wir uns hier auf die Bestimmung des gesamten Repertoires der SNCA-Transkript-Isoformen bei verschiedenen Synucleinopathien konzentriert. Zuvor wurden für das SNCA-Gen mehrere verschiedene SNCA-Transkript-Isoformen beschrieben, die durch alternatives Spleißen, Transkriptionsstartstellen (TSS) und die Auswahl von Polyadenylierungsstellen entstanden sind (McLean et al., 2012; Xu et al., 2014). Alternatives Spleißen der kodierenden Exons führt zu SNCA 140, SNCA 112, SNCA 126 und SNCA 98, was zu vier Protein-Isoformen führt (Beyer und Ariza, 2012). Alternative TSSs des SNCA-Gens führen zu vier verschiedenen 5′UTRs, und die alternative Auswahl verschiedener Polyadenylierungsstellen bestimmt drei Hauptlängen der 3′UTR, ohne Auswirkungen auf die Zusammensetzung des Proteinprodukts (Beyer und Ariza, 2012). Unser übergeordnetes Ziel ist es, neue Erkenntnisse über den Beitrag der verschiedenen bekannten und neuen SNCA-mRNA-Spezies zur Pathogenese und Heterogenität von Synucleinopathien zu gewinnen.
Bislang haben die meisten Studien Short-Read-Sequenzierungstechnologien verwendet, um die Transkriptomkomplexität in menschlichen Gehirnen zu erforschen. Die Verfügbarkeit von Long-Read-Technologien der dritten Generation bietet ein noch nie dagewesenes und nahezu vollständiges Bild der Isoformstrukturen. Bei der bisherigen Long-Read-Transkript-Sequenzierung für menschliche Krankheitsgene wurde jedoch ein Amplikon-basierter Ansatz verwendet (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Dieser Ansatz hat sich zwar bei der Identifizierung von komplexem alternativem Spleißen in menschlichen Krankheitsgenen als erfolgreich erwiesen, ist jedoch auf das Design der PCR-Primer beschränkt und deckt keine alternativen Start- und Endstellen auf. Gezielte Anreicherung, z. B. durch die Verwendung von IDT-Sonden, kann bei geringen Sequenzierungskosten einen umfassenden Überblick über die Isoformen von Genen von Interesse liefern. Darüber hinaus ermöglichen hochpräzise Transkript-Reads in voller Länge eine isoformspezifische Haplotypisierung.
Hier stellen wir die erste bekannte Studie vor, in der die gDNA und cDNA der SNCA-Genregion mit PacBio SMRT-Sequenzierung gezielt erfasst wurde. Die SNCA-Genregion ist ~114 kb lang und besteht aus sechs Exons mit Transkriptlängen um 3 kb. Wir haben 12 menschliche Gehirnproben von Morbus Parkinson, DLB und normalen Kontrollproben gemultiplext und die gDNA- und cDNA-Bibliothek mit dem PacBio Sequel System sequenziert. Wir beschreiben die bioinformatischen Analysen zur Identifizierung von SNPs, Indels und kurzen Tandemwiederholungen für die gDNA-Erfassung und die Haplotypisierung auf Isoformebene für die cDNA-Daten. Wir zeigen, dass die gezielte Erfassung eine kosteneffiziente Methode zur gemeinsamen Untersuchung der genomischen Variation und des alternativen Spleißens in einem krankheitsbezogenen neuronalen Gen ist.
Materialien und Methoden
Studienproben
Die Studienkohorte (N = 12) bestand aus Personen mit drei autopsiebestätigten neuropathologischen Diagnosen: (1) Morbus Parkinson (N = 4); (2) DLB (N = 4); und (3) klinisch und neuropathologisch normalen Personen (N = 4). Das Hirngewebe des Frontalkortex wurde von der Kathleen Price Bryan Brain Bank (KPBBB) der Duke University, dem Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) und dem Layton Aging and Alzheimer’s Disease Center der Oregon Health and Science University zur Verfügung gestellt. Die neuropathologischen Phänotypen wurden bei der Postmortem-Untersuchung nach etablierten Standardmethoden bestimmt, die den Methoden und klinischen Praxisempfehlungen von McKeith und Kollegen folgen (McKeith et al., 1999, 2005). Die Dichte der LB-Pathologie (in einem Standardsatz von Hirnregionen) wurde mit den Noten leicht, mittelschwer, schwer und sehr schwer bewertet. Die Studienproben innerhalb jeder Diagnosegruppe, PD und DLB, wurden sorgfältig ausgewählt, so dass der Schweregrad der klinisch-pathologischen Phänotypen innerhalb jeder Pathologie ähnlich war. Alle Gehirne wiesen Lewy-Körperchen im Hirnstamm, im Limbus und in der Neokortikalis auf, während Morbus Parkinson schwere bis sehr schwere McKeith-Scores in der Subnigra und der Amygdala zeigte. Alle Gehirne wiesen nach CERAD-Kriterien und Braak und Braak-Stadium = II keine AD auf. Die neurologisch gesunden Hirnproben wurden aus postmortalem Gewebe klinisch unauffälliger Probanden gewonnen, die in den meisten Fällen innerhalb eines Jahres nach dem Tod untersucht wurden und bei denen keine kognitiven Störungen oder Parkinsonismus sowie neuropathologische Befunde festgestellt wurden, die nicht für die Diagnose von Parkinson, Alzheimer oder anderen neurodegenerativen Erkrankungen ausreichten. Alle Stichproben waren Weiße. Die demografischen Daten und die Neuropathologie dieser Probanden sind in der ergänzenden Tabelle 1 zusammengefasst. Das Projekt wurde vom Duke Institution Review Board (IRB) genehmigt, das auch die ethische Genehmigung erteilte. Die Methoden wurden in Übereinstimmung mit den einschlägigen Richtlinien und Vorschriften durchgeführt.
Genomische DNA- und RNA-Extraktionen
Genomische DNA wurde aus Hirngewebe nach dem Standardprotokoll von Qiagen (Qiagen, Valencia, CA) extrahiert. Die Gesamt-RNA wurde aus Hirnproben (100 mg) mit TRIzol-Reagenz (Invitrogen, Carlsbad, CA) extrahiert und anschließend mit einem RNeasy-Kit (Qiagen, Valencia, CA) nach dem Protokoll des Herstellers gereinigt. Die gDNA- und RNA-Konzentration wurde spektrophotometrisch bestimmt, und die Qualität der RNA-Proben und das Fehlen eines signifikanten Abbaus wurden durch Messungen der RNA-Integritätszahl (RIN, ergänzende Tabelle 1) mit einem Agilent Bioanalyzer bestätigt.
Bibliotheksvorbereitung und Sequenzierung
gDNA-Capture mit IDT Xgen® Lockdown®-Sonden und Einzelmolekül-Sequenzierung
Ungefähr 2 μg jeder gDNA-Probe wurden mit dem Covaris g-TUBE auf 6 kb geschert und mit barcodierten Adaptern ligiert. Ein äquimolarer Pool von 12-fach barcodierter gDNA-Bibliothek (insgesamt 2 μg) wurde in die sondenbasierte Erfassung mit einem speziell entwickelten SNCA-Gen-Panel eingegeben.
Eine SMRTBell-Bibliothek wurde unter Verwendung von 626 ng erfasster und reamplifizierter gDNA1 erstellt.
CDNA-Capture mit IDT Xgen® Lockdown®-Sonden und Einzelmolekül-Isoform-Sequenzierung (Iso-Seq)
Ungefähr 100-150 ng Gesamt-RNA pro Reaktion wurden mit dem Clontech SMARTer cDNA-Synthese-Kit und 12 probenspezifischen barcodierten Oligo dT (mit PacBio 16mer Barcode-Sequenzen, siehe ergänzende Methoden) revers transkribiert. Für jede Probe wurden drei Reaktionen der reversen Transkription (RT) parallel durchgeführt. Die PCR-Optimierung diente zur Bestimmung der optimalen Amplifikationszykluszahl für die nachgeschalteten groß angelegten PCR-Reaktionen. Ein einziger Primer (Primer IIA aus dem Clontech SMARTer Kit 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) wurde für alle PCR-Reaktionen nach der RT verwendet. PCR-Produkte im großen Maßstab wurden separat mit 1X AMPure PB-Beads gereinigt, und der Bioanalyzer wurde zur Qualitätskontrolle verwendet. Ein äquimolarer Pool von 12-plex barcodierten cDNA-Bibliotheken (insgesamt 1 μg) wurde in die sondenbasierte Erfassung mit einem speziell entwickelten SNCA-Gen-Panel eingegeben.
Eine SMRTBell-Bibliothek wurde unter Verwendung von 874 ng erfasster und reamplifizierter cDNA2 erstellt. Eine SMRT-Zelle 1M (6 Stunden Film) wurde auf der PacBio Sequel-Plattform unter Verwendung von 2.0-Chemie sequenziert.
gDNA-Analyse
Die Sequenzierung der barcodierten gDNA-Daten wurde an drei SMRT-Zellen 1M unter Verwendung von 2.0-Chemie durchgeführt. Die Daten wurden demultiplexiert, indem die Anwendung Demultiplex Barcodes in PacBio SMRT Link v6.0 ausgeführt wurde.
Kurzvariantenanalyse und Phasing
Circular Consensus Sequence (CCS) Reads wurden mit SMRT Analysis 6.0 aus jedem demultiplexierten Datensatz generiert und mit minimap2 an das hg19-Referenzgenom angeglichen. PCR-Duplikate aus der Post-Capture-Amplifikation wurden durch Mapping der Endpunkte identifiziert und mit einem benutzerdefinierten Skript markiert. Kurze Varianten wurden mit GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018) aufgerufen. Nach einem ersten Durchgang der Filterung mit Abdeckungstiefe und Qualitätsmetriken wurden die Varianten manuell in IGV3 inspiziert. Wenn Varianten nicht mit nahegelegenen SNPs übereinstimmten, wurden sie manuell gefiltert. Die Variantenstellen, die die manuelle Kuration bestanden, wurden in Verbindung mit den deduplizierten CCS-Alignments für Read-Backed-Phasing mit WhatsHap (Martin et al., 2016) verwendet.
Clustering und Bestimmung von Haplotypen für CT-Rich Region
Subsequenzen, die auf chr4: 90742331-90742559 (hg19) ausgerichtet waren, wurden für jede Probe extrahiert. Nach Prüfung der Größenverteilung dieser Teilsequenzen wurden sie mit Hilfe einer Kombination aus Python und MUSCLE (Edgar, 2004) nach Größe und Sequenzähnlichkeit geclustert, und für jeden Cluster wurde unabhängig eine Konsenssequenz erstellt.
Benutzerdefinierte Skripte und Arbeitsabläufe werden in https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases weiter beschrieben.
Isoform-Analyse
Die Sequenzierung der barcodierten cDNA-Daten erfolgte auf einer SMRT Cell 1M auf dem PacBio Sequel System unter Verwendung von 2.0 Chemie. Die bioinformatische Analyse wurde mit der IsoSeq3-Anwendung in der PacBio SMRT Analysis v6.0.0 durchgeführt, um qualitativ hochwertige Isoform-Sequenzen in voller Länge zu erhalten (siehe ergänzende Methoden für weitere Informationen).
Isoform SNP Calling
Die mit den endgültigen 41 Isoformen aus allen 12 Proben assoziierten Isoform-Reads in voller Länge wurden an das hg19-Genom angeglichen, um einen Pileup zu erstellen. Basen mit einem QV von weniger als 13 wurden ausgeschlossen. Anschließend wurde an jeder Position mit einer Abdeckung von mindestens 40 Basen ein exakter Fisher-Test mit Bonferroni-Korrektur und einem p-Cutoff von 0,01 durchgeführt. Nur Substitutions-SNPs, die nicht in der Nähe von homopolymeren Regionen (Abschnitte mit 4 oder mehr gleichen Nukleotiden) liegen, wurden ermittelt. Nach dem SNP-Calling wurde der Genotyp für jede Probe bestimmt, indem die Anzahl der unterstützenden probenspezifischen Full Length (FL) Reads gezählt wurde. Wenn eine Probe 5+ FL-Reads aufwies, die sowohl die Referenz- als auch die alternative Base unterstützten, war sie heterozygot. Wenn eine Probe 5+ FL-Reads für ein Allel und weniger als 5 FL-Reads für das andere Allel aufwies, war sie homozygot. Andernfalls war sie nicht schlüssig. Die Skripte sind verfügbar unter: https://github.com/Magdoll/cDNA_Cupcake.
Ergebnisse
Wir entwarfen benutzerdefinierte Sonden für das SNCA-Gen und führten eine gezielte Erfassung von gDNA und cDNA in einer Multiplex-Bibliothek durch, die aus 12 menschlichen Gehirnproben von Morbus Parkinson, DLB und normalen Kontrollen bestand (Abbildung 1, ergänzende Tabelle 1). Die gDNA- und cDNA-Bibliotheken wurden mit der PacBio Sequel-Plattform sequenziert. Die bioinformatische Analyse erfolgte mit PacBio-Software, gefolgt von einer benutzerdefinierten Analyse.
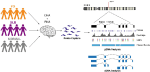
Abbildung 1. Schematische Darstellung des Studiendesigns. DNA- und RNA-Materialien wurden aus postmortalem Hirngewebe von Patienten mit Morbus Parkinson, Demenz mit Lewy-Körperchen und Kontrollgruppen extrahiert. gDNA- und cDNA-Bibliotheken wurden durch Sondenhybridisierung hergestellt und mit dem PacBio Sequel System sequenziert. Die Analyse wurde mit PacBio-Software und anderen vorhandenen Tools durchgeführt.
Gezieltes gDNA-Capture identifizierte bekannte und neue Variationen
Nach der Generierung zirkulärer Konsensussequenzen (CCS) und der Entfernung von PCR-Duplikaten (ergänzende Methoden) erhielten wir eine 16- bis 71-fache mittlere einzigartige Abdeckung der SNCA-Genregion. Die CCS-Reads hatten eine mittlere Insertlänge von 2,9 kb und eine mittlere Lesegenauigkeit von 98,9%. Mit Ausnahme einer 5-kb-Region, die aufgrund des Vorhandenseins von LINE-Elementen (hg19 chr4: 90697216-90702113) und einer 2,1-kb-Region mit hohem GC-Gehalt um Exon 1 absichtlich von den Sonden nicht abgedeckt wurde, gab es eine ausreichende Abdeckung, um beide Haplotypen für jede der 212 Proben zu genotypisieren (Abbildung 2, ergänzende Abbildung 1).

Abbildung 2. Gezielter gDNA-Einfang und Phasierung. Ein Beispiel mit einer Probe aus jeder Bedingung. Die obere Spur zeigt eine der SNCA-Isoformen, gefolgt von der gDNA-Abdeckung für die drei Proben. Die Variantenspur zeigt jeden SNP und ist farbkodiert für heterozygot (lila), homozygot alternativ (orange) und homozygot Referenz (grau). Phasierte Blöcke sind in Hellblau dargestellt. Die untere Spur zeigt die Positionen der Capture-Sonden. Die Dropout-Region im Sondendesign ist auf zwei LINE-Elemente in der Mitte von Intron 4 zurückzuführen. Für die gDNA-Abdeckung und Phasing-Informationen aller 12 Proben siehe ergänzende Abbildungen.
Mit Hilfe von GATK4 HC, qualitätsbasierter Filterung und manueller Kuration identifizierten wir 282 SNPs und 35 Indels, einschließlich 8 SNPS und 13 Indels, die nicht in dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) gefunden wurden (ergänzende Tabelle 2). In der kodierenden Region für SNCA wurden keine Varianten identifiziert, allerdings wurden acht Varianten in den untranslatierten Regionen gefunden. Die Mehrzahl der identifizierten Varianten, darunter mehrere kurze Tandem-Repeats (STR), befinden sich in den Introns 2, 3 und 4.
Wir haben zuvor eine hoch polymorphe CT-reiche Region in Intron 4 von SNCA mit vier beobachteten Haplotypen beschrieben (Lutz et al., 2015). Während sich diese hoch repetitive und strukturell variable Region als schwierig erwies, mit GATK4 HC zu genotypisieren, waren wir in der Lage, Konsenssequenzen für alle 12 Proben zu konstruieren und beobachteten alle 4 der zuvor entdeckten Haplotypen (ergänzende Abbildung 2). Zusätzlich identifizierten wir eine neue STR in Intron 4, die aus einer Drei-Basen-Einheit besteht, die in der Referenz 16 Mal wiederholt wird. Innerhalb der 12 Proben identifizierten wir drei Haplotypen mit 9, 12 und 15 Kopien der TTG-Wiederholungseinheit. GATK HC hat alle diese Haplotypen korrekt genotypisiert, mit Ausnahme eines Haplotypen für PD-4, der in dieser Region eine relativ geringe Abdeckung aufwies. Mit den vorliegenden Daten für diese Probe kann der Genotyp jedoch durch visuelle Inspektion bestimmt werden (Tabelle 1).

Table 1. Ein neuartiges Triplett-Tandem-Repeat in Intron 4 (chr4: 90713442).
Wir haben die kurzen Varianten, die von GATK HC erkannt wurden, in Verbindung mit dem Read-basierten Phasing-Tool WhatsHap (Martin et al., 2016) verwendet, um die CCS-Reads über den Locus zu phasen, mit einer Reihe von Erfolgen, die hauptsächlich von der heterozygoten Variantendichte über den Locus bestimmt wurden. Die Proben PD-1, PD-4, N-4, DLB-1 und DLB-4 wiesen lange Abschnitte mit geringer Heterozygotie und sehr wenigen, kurzen Phasenblöcken auf, während die anderen Proben Phasenblöcke im Bereich des 7- bis 18-fachen der mittleren Leselänge, bis zu 54 kb, ergaben (ergänzende Abbildung 3).
Targeted cDNA Capture Identified Novel Start and End Sites
Wir verarbeiteten die PacBio cDNA (Iso-Seq) Daten mit der PacBio SMRT Analysis Software. Nach dem Mapping der Iso-Seq-Daten auf hg19 und der Entfernung von Artefakten (ergänzende Tabelle 3, ergänzende Abbildung 4) erhielten wir einen endgültigen Satz von 41 SNCA-Isoformen (Abbildung 3). Alle endgültigen Isoformen haben alle kanonischen Spleißstellen (GT-AG oder GC-AG) und werden durch insgesamt 20 oder mehr Volllängen-Reads unterstützt. Die Mehrheit der Isoformen (28 von 41) hat alle sechs Exons und unterscheidet sich nur durch die Verwendung alternativer 5′-Startstellen und 3′-UTR-Längen. Die 3′ UTR-Längen variierten zwischen 300 und 2,6 kb. Die Verwendung sehr unterschiedlicher alternativer 5′-Startstellen in SNCA ist bekannt; weniger bekannt ist die variable 3′-UTR-Länge, die zuvor anhand von RNA-seq-Daten untersucht worden war, die keine Isoform-Strukturen in voller Länge auflösten (Rhinn et al., 2012). Die Iso-Seq-Daten zeigen, dass die variable 3′-UTR-Länge mit allen möglichen Kombinationen von 5′-Startstellen gepaart zu sein scheint, ohne dass eine bevorzugte Kopplung vorliegt. Fast nichts von der Variabilität der Start- und Endstellen verändert das vorhergesagte offene Leseraster (ergänzende Abbildung 5) und wird voraussichtlich in die kanonische 141-Aminosäuresequenz übersetzt.

Abbildung 3. Mit gezielter Iso-Seq erfasste SNCA-Isoformen identifizieren neue Start- und Endstellen. Der Großteil der Isoformkomplexität ist auf die kombinatorische Verwendung alternativer 3′ UTR-Längen und Exon 1 zurückzuführen, wobei einige seltene alternative Spleißstellen in Exon 1 (grün), 2 (rot) und 4 (blau) gefunden wurden. Alle Verbindungsstellen haben kanonische Spleißstellen. Wir identifizierten fünf Isoformen, die Exon 5 skippen, und zwei Isoformen, die Exon 3 skippen. Wir haben auch neue Start- (orange) und Endstellen (lila) in Intron 4 identifiziert. Aufgerufene SNPs sind lila markiert.
Wir haben die neuartigen (aber kanonischen) Kreuzungen anhand öffentlich zugänglicher Short-Read-Kreuzungsdaten weiter validiert. Die Intropolis-Datenbank (v1, https://github.com/nellore/intropolis) vereint über 21.000 öffentlich verfügbare RNA-seq-Daten. Aufgrund des hohen Volumens an Kreuzungsdaten, die nur durch einen einzigen Short-Read unterstützt werden, benötigen wir für diese Studie mindestens 10 Short-Read-Unterstützungen (kombiniert aus allen >21.000 RNA-seq-Datensätzen), um unsere neuen Iso-Seq-Kreuzungen zu bestätigen. Mit Ausnahme der neuen Verbindungen für PB.1016.253 und PB.1016.296 (Abbildung 3) werden alle anderen neuen Verbindungen durch den Intropolis-Datensatz unterstützt. Interessanterweise haben diese neuartigen Verbindungen eine deutlich geringere Unterstützung durch kurze Lesungen als die mit Gencode annotierten Verbindungen. Zum Beispiel werden die beiden neuen Verbindungen in PB.1016.139, die durch das neue Exon eingeführt wurden, durch 2.519 bzw. 44 Intropolis-Kurzlesezahlen unterstützt, während die anderen vier bekannten Verbindungen durch über 1 Million Kurzlesezahlen unterstützt werden. Dies zeigt, wie leistungsfähig die gezielte Anreicherung mit Hilfe der Transkriptomsequenzierung in voller Länge ist, um seltene, neuartige Isoformen zu entdecken.
Wir beobachteten zwei Isoformen mit Exon 3-Skipping (SNCA126) und fünf Isoformen mit Exon 5-Skipping (SNCA112). Auch hier ist die Spleißvielfalt in diesen beiden Exon-Skipping-Gruppen hauptsächlich auf die unterschiedliche Verwendung alternativer 5′-Startstellen und die variable 3′-UTR-Länge zurückzuführen. Die ORF-Vorhersage zeigt, dass das Skippen von Exon 3 oder Exon 5 den ORF verkürzt, aber das Leseraster beibehält. Drei Isoformen haben neue 3′-Endstellen in Intron 4. Die ORF-Vorhersage zeigt, dass dies zu einem verkürzten Proteinprodukt führt.
Wir identifizierten eine bisher nicht annotierte 5′-Startstelle in Intron 4 (hg19 chr4: 90692548-90693045, Abbildung 3). Die drei Isoformen, die mit diesem neuen Startpunkt assoziiert sind, bestehen aus dem neuen Startpunkt, Exon 5 und variablen 3′ UTR-Längen. Interessanterweise, während öffentlich heruntergeladene Short-Read-Daten von GTEx und Sandor et al. (2017) und CAGE-Peak-Daten (FANTOM5) diese neuartige Startstelle nicht unterstützen, enthielt ein kürzlich veröffentlichter NA12878-Direkt-RNA-Datensatz4 nur ein SNCA-Transkript, das diese alternative Startstelle bestätigt. Darüber hinaus wird die neuartige Verbindung zwischen Exon 5 und der neuartigen Startstelle durch Intropolis Short-Read-Verbindungsdaten bestätigt. Interessanterweise wird diese neue 5′-Startstelle für die Einführung neuer Peptide vorhergesagt, während das Leseraster in Exon 5 beibehalten wird.
Wir identifizierten auch drei SNCA-Transkripte mit neuen Endstellen (Abbildung 3). Zwei Isoformen (PB.1016.383, PB.1016.384) verwendeten eine verlängerte 3′ UTR in Exon 4, während die dritte Isoform (PB.1016.381) ein neuartiges 3′ Exon in Intron 4 verwendete. Die neuartigen Verbindungen zwischen dem neuartigen letzten Exon und dem vorherigen Exon werden durch öffentliche Short-Read-Verbindungsdaten (Intropolis) bestätigt. Die neuartigen 3′ UTRs führen zu einer verkürzten ORF-Vorhersage.
Wenn man die normalisierte Anzahl der Volllängen-Lesungen als Proxy für die Isoform-Häufigkeit verwendet, stellt man fest, dass eine der kanonischen SNCA-Isoformen (PB.1016.131) am häufigsten vorkommt, mit einer Häufigkeit von 50-60% in allen untersuchten Proben (ergänzende Tabelle 4). Wir gruppierten die 41 Isoformen weiter nach ihren Spleißmustern (Tabelle 2). Isoformen, die alle sechs Exons aufweisen, machen 95-97 % der Häufigkeit aus. Frühere Studien haben einen deutlichen Anstieg der Expression von Isoformen, denen Exon 3 fehlt (SNCA126), im frontalen Kortex von DLB-Proben im Vergleich zu normalen Proben gezeigt (Beyer et al., 2008); unsere aggregierten Isoformzahlen zeigen, dass drei der DLB-Proben ein leicht erhöhtes Zählniveau im Vergleich zu den normalen Proben aufweisen, ebenso wie die SNCA112-Varianten (Exon 5-Skipping) für PD und DLB im Vergleich zu normalen Proben.

Table 2. SNCA-Isoform-Häufigkeit für jede Probe, aggregiert nach Spleißmustern.
CDNA in voller Länge ermöglicht Phasing-Informationen auf Isoform-Ebene
Wir riefen SNPs unter Verwendung von cDNA auf, indem wir alle Reads in voller Länge aus den 12 Proben anhäuften, um Varianten zu rufen (siehe Abschnitt “Methoden”). Insgesamt wurden vier SNPs genannt, die alle zuvor in dbSNP annotiert worden waren (Tabelle 3, Abbildung 3). Die vier SNPs befinden sich alle in Nicht-CDS-Regionen, einer in der 3′ UTR (Exon 6), einer im Intron 4 und zwei in der 5′ UTR (Exon 1). Der 3′ UTR SNP (chr4: 90646886) wird nur von Isoformen mit einem 3′ UTR abgedeckt, der mindestens ~1 kb lang ist, und daher decken nicht alle kanonischen Isoformen diesen SNP ab. Der Intron-4-SNP (chr4: 90743331) wird nur von den neuartigen alternativen 3′-End-Isoformen (PB.1016.383, PB.1016.384) abgedeckt und ist mit keinem der anderen SNPs verbunden. Die beiden 5′-UTR-SNPs (chr4: 90757312 und chr4: 90758389) werden von zwei sich gegenseitig ausschließenden Exon-1-Nutzungen abgedeckt und sind daher ebenfalls nicht verknüpft.

Tabelle 3. cDNA-SNP-Informationen.
Unser derzeitiger Ansatz beschränkt sich darauf, nur Substitutionsvarianten in transkribierten Regionen mit ausreichender Abdeckung zu nennen. Ein Vergleich der Liste unserer SNPs mit der hg19 dbSNP-Annotation zeigt, dass die meisten der übersehenen SNPs oder Varianten entweder eine Häufigkeit von weniger als 1 % in der Bevölkerung aufweisen, keine Einzelnukleotidsubstitutionen sind oder an Regionen mit geringer Komplexität angrenzen. So wird beispielsweise rs77964369 (chr4: 90646532) mit einer 50/50-Häufigkeit von T/A angegeben; dieses T liegt jedoch neben einem Abschnitt von 11 genomischen As stromabwärts. Die manuelle Inspektion des Iso-Seq-Read-Pileups, das an dieser Stelle ~1.300 Reads aufweist, deutet nicht auf eine Variation hin, zumindest nicht bei unseren 12 Proben.
Unter Verwendung der probenspezifischen Reads rufen wir den Genotyp jeder Probe an jeder SNP-Position auf (Tabelle 3). Abgesehen von PD-2, das zu wenige Reads hat und für alle vier SNPs nicht schlüssig ist, konnten wir den Genotyp für die meisten anderen Proben bestimmen. Bemerkenswert ist, dass DLB-3 die einzige Probe war, die an allen SNP-Positionen heterozygot ist. Ansonsten konnten wir kein krankheitsspezifisches Muster der Bevorzugung eines Genotyps gegenüber einem anderen beobachten.
Diskussion
Wir beschreiben die erste Studie, in der eine gezielte Anreicherung des SNCA-Gens in multiplexen gDNA- und cDNA-Bibliotheken zur Untersuchung neurologischer Erkrankungen mittels Long-Read-Sequenzierung eingesetzt wurde. Die langen Leselängen des PacBio Sequel Systems erleichterten die Sequenzierung des gesamten Repertoires an Transkript-Isoformen des SNCA-Gens. Wir haben die Vielfalt in der Verwendung alternativer 5′-Startstellen und variabler 3′-UTR-Längen aufgedeckt und bekannte Exon-Skipping-Ereignisse beobachtet, wie die Deletion von Exon 3 (SNCA126) und Exon 5 (SNCA112). Darüber hinaus wurden neue alternative Start- und Endstellen innerhalb des großen Intron 4 identifiziert, von denen man annimmt, dass sie in neue Proteine übersetzt werden. Es ist wahrscheinlich, dass die hohe Sequenzierungstiefe der gezielten Erfassung in Kombination mit der Fähigkeit, komplette Transkripte zu sequenzieren, es uns ermöglichte, diese bisher unbeschriebenen Isoformen zu entdecken.
Die biologische und pathologische Bedeutung der verschiedenen SNCA-Protein-Isoformen muss noch vollständig entdeckt werden. Bestimmte SNCA-Isoformen, die nach der Translation modifiziert und gespleißt werden, wurden jedoch mit intrazellulären Aggregationsneigungen in Verbindung gebracht (Kalivendi et al., 2010) und werden bei menschlichen Synucleinopathien unterschiedlich exprimiert (Beyer et al., 2008; Beyer und Ariza, 2012). Studien zur posttranslationalen Modifikation von SNCA zeigten, dass Lewy-Körperchen, das pathologische Merkmal von Synucleinopathien, reichlich phosphoryliertes, nitriertes und monoubiquitiniertes SNCA enthalten (Kim et al., 2014). Die Auswirkungen der posttranskriptionellen Modifikation auf die SNCA-Aggregation wurden ebenfalls untersucht. Es wurde vermutet, dass alternatives Spleißen die SNCA-Aggregation beeinflusst. Eine Deletion von Exon 3 oder 5 hat funktionelle Konsequenzen: Die Deletion von Exon 3 (SNCA126) führt zur Unterbrechung der N-terminalen Protein-Membran-Interaktionsdomäne, was zu einer geringeren Aggregation führen kann, und die Deletion von Exon 5 (SNCA112) kann aufgrund einer deutlichen Verkürzung des unstrukturierten C-Terminus zu einer verstärkten Aggregation führen (Lee et al., 2001; Beyer, 2006). Im frontalen Kortex von DLB-Patienten ist SNCA112 im Vergleich zu Kontrollpersonen deutlich erhöht (Beyer et al., 2008), während die SNCA126-Spiegel im präfrontalen Kortex von DLB-Patienten verringert sind (Beyer et al., 2006). Im Gegensatz dazu zeigte sich eine erhöhte SNCA126-Expression im frontalen Kortex von Parkinson-Gehirnen und keine signifikanten Unterschiede bei MSA (Beyer et al., 2008). SNCA98 ist eine hirnspezifische Spleißvariante, der sowohl Exon 3 als auch 5 fehlen und die in verschiedenen Bereichen des fötalen und erwachsenen Gehirns unterschiedliche Expressionsniveaus aufweist. Eine Überexpression von SNCA98 wurde in den Frontalkortexen von DLB, PD (Beyer et al., 2007) und MSA (Beyer et al., 2008) im Vergleich zu Kontrollen festgestellt. Darüber hinaus wurde berichtet, dass der posttranskriptionelle Prozess, der zu einer alternativen 3′UTR-Nutzung führt, Auswirkungen auf die mRNA-Stabilität und -Lokalisierung hat (Fabian et al., 2010; Rhinn et al., 2012; Yeh und Yong, 2016). Weitere Untersuchungen hinsichtlich der Aggregationsneigung der verschiedenen bekannten SNCA-Protein-Isoformen und der Zusammensetzung der Lewy-Körperchen sind gerechtfertigt. Darüber hinaus legte unsere Studie den Grundstein für mRNA-Quantifizierungsanalysen der bisher bekannten und neuartigen Transkripte in einer größeren Stichprobe von Probanden mit einer Reihe von klinisch-pathologischen Stadien unter Verwendung mehrerer Hirnregionen von jedem Probanden. Diese Analysen der hirnregionsspezifischen transkriptomischen Landschaft von SNCA im Kontext des neuropathologischen Schweregrads werden aufschlussreich sein im Hinblick auf die Rolle spezifischer SNCA-Transkript-Isoformen beim Fortschreiten der neuropathologischen Stadien und der Schwere der Lewy-Körperchen und der Lewy-Neuritendichte.
In dieser Arbeit haben wir uns auf die Schaffung eines Sequenzierungs- und Analysestandards für die Analyse gezielter gDNA- und cDNA-Daten konzentriert, die von denselben Probanden generiert wurden. Dies ist ein leistungsfähiger Ansatz, der es ermöglicht, die gDNA-Sequenzen über die gesamte Region eines bestimmten Gens auf der Grundlage der Heterozygotie in der Sequenz der Isoformen des Volllängentranskripts zu ordnen. Die PacBio-gezielten gDNA-Daten in dieser Studie ergaben phasierte Blöcke, die 81 % der 114 kb großen Region um SNCA abdeckten, wobei der längste phasierte Block 54 kb überstieg. Da die gDNA-Phasierung durch die Leselänge und die Heterozygotie begrenzt ist, werden mit zunehmender Leselänge wahrscheinlich größere Phasenblöcke erzeugt.
Die gDNA-Variantenanalyse bestätigte bekannte und identifizierte neue Short Tandem Repeats (STRs) in den intronischen Regionen. Beispielsweise entdeckten wir zuvor mithilfe von Phasensequenzierung durch Klonierung und Sanger-Sequenzierung vier unterschiedliche Haplotypen innerhalb einer intronischen CT-reichen Region, die aus einem Cluster variabler repetitiver Sequenzen bestand (Lutz et al., 2015). Wir konnten zeigen, dass ein spezifischer Haplotyp, der sogenannte Haplotyp 3, das Risiko für die Entwicklung der Lewy-Körperchen-Pathologie bei Alzheimer-Patienten erhöht. Hier haben wir die Sequenz dieser hoch polymorphen Region mit geringer Komplexität und ihre vier definierten Haplotypen validiert. Obwohl unsere Stichprobengröße klein war, war der Haplotyp 3″ ausschließlich bei Patienten mit Alzheimer-Krankheit (ein Morbus-Parkinson-Patient, zwei DLB-Patienten) vorhanden, was mit unseren früheren Ergebnissen übereinstimmt. Die Pilotergebnisse und unsere frühere Veröffentlichung bilden die Grundlage für eine Wiederholung der Assoziationsanalysen von Synucleinopathien mit genau definierten, d. h. durch Long Reads bestimmten STRs und strukturellen Haplotypen unter Verwendung einer größeren Stichprobengröße.
Unsere Arbeit hat gezeigt, dass das PacBio Sequel System in der Lage ist, neue Transkripte in voller Länge zu entdecken und das gesamte Repertoire an Transkripten in voller Länge eines Gens zu charakterisieren, das an einer Krankheit beteiligt ist. Darüber hinaus haben wir gezeigt, dass Long Reads gDNA kurze strukturelle Varianten und Haplotypen, einschließlich STRs, genauer definieren und dadurch die Entdeckung und Validierung anderer krankheitsassoziierter Varianten als SNPs erleichtern können. Insgesamt sind diese neuen Erkenntnisse sehr wertvoll und können dazu beitragen, unser Verständnis der genetischen Ursachen komplexer menschlicher Krankheiten, einschließlich altersbedingter neurodegenerativer Erkrankungen wie Synucleinopathien, zu verbessern, die möglicherweise mit Störungen in der Transkriptlandschaft einhergehen.
Datenverfügbarkeit
Die drei SMRT-Zellen der gDNA-Rohdaten sind unter Zenodo.org mit doi: 10.5281/zenodo.1560688 verfügbar. Die eine SMRT-Zelle der cDNA-Rohdaten ist bei Zenodo.org unter doi: 10.5281/zenodo.1581809 zu finden. Die bearbeiteten gDNA- und cDNA-Ergebnisse, einschließlich gDNA-Varianten und cDNA-Isoformen, sind unter Zenodo.org mit doi: 10.5281/zenodo.3261805.
Beiträge der Autoren
OC-F trug zur Konzeption und Gestaltung der Studie bei. ET und WR organisierten Sequenzdatenbanken, führten die Sequenzierungsanalysen durch und erstellten alle Abbildungen und Tabellen. O-CG und JB kümmerten sich um die Hirngewebe- und Nukleinprobenpräparate. TH erstellte die Sequenzierungsdatensätze. SK entwarf und beschaffte die Reagenzien. OC-F, ET und WR verfassten den ersten Entwurf des Manuskripts. OC-F erhielt finanzielle Unterstützung. Alle Autoren trugen zur Vorbereitung des Manuskripts bei, lasen und genehmigten die eingereichte Version.
Finanzierung
Diese Arbeit wurde zum Teil von den National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) finanziert.
Erklärung zu Interessenkonflikten
ET, WR, TH und SK sind oder waren zum Zeitpunkt der Studie Mitarbeiter von Pacific Biosciences.
Die übrigen Autoren erklären, dass die Forschung in Abwesenheit jeglicher kommerzieller oder finanzieller Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
Danksagungen
Dieses Manuskript wurde als Preprint bei BioRxiv veröffentlicht (Tseng et al., 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Ergänzendes Material
Das ergänzende Material zu diesem Artikel finden Sie online unter: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona-Studie zu Alterung und neurodegenerativen Erkrankungen und Gehirn- und Körperspendenprogramm. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-Synucleinstruktur, posttranslationale Modifikation und alternatives Spleißen als Aggregationsverstärker. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Alpha-Synuclein posttranslationale Modifikation und alternatives Spleißen als Auslöser für Neurodegeneration. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., and Ariza, A. (2008). Unterschiedliche Expression der Isoformen von Alpha-Synuclein, Parkin und Synphilin-1 bei der Lewy-Körperchen-Krankheit. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Identifizierung und Charakterisierung einer neuen Alpha-Synuclein-Isoform und ihre Rolle bei Lewy-Körperchen-Erkrankungen. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Niedrige Alpha-Synuclein-126-mRNA-Spiegel bei Demenz mit Lewy-Körperchen und Alzheimer-Krankheit. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., und Filipowicz, W. (2010). Regulation der mRNA-Translation und -Stabilität durch microRNAs. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., und Kalyanaraman, B. (2010). Oxidantien induzieren alternatives Spleißen von Α-Synuclein: Implikationen für die Parkinson-Krankheit. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., and Halliday, G. M. (2014). Die Biologie von Alpha-Synuclein bei Lewy-Körperchen-Erkrankungen. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgenrezeptor-Variante AR-V9 wird mit AR-V7 in Prostatakrebsmetastasen koexprimiert und sagt Abirateron-Resistenz voraus. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). Membrangebundenes Α-Synuclein hat eine hohe Aggregationsneigung und die Fähigkeit, die Aggregation der zytosolischen Form zu fördern. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., and Chiba-Falek, O. (2015). Ein Cytosin-Thymin (CT)-reicher Haplotyp in Intron 4 von SNCA ist mit einem Risiko für Lewy-Körper-Pathologie bei Alzheimer verbunden und beeinflusst die SNCA-Expression. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnose und Behandlung der Demenz mit Lewy-Körperchen: Dritter Bericht des DLB-Konsortiums. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Bericht über den zweiten internationalen Workshop zur Demenz mit Lewy-Körperchen: Diagnose und Behandlung. Konsortium für Demenz mit Lewy-Körperchen. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., and Isacson, O. (2012). Transkriptionsniveaus von Alpha-Synuclein in voller Länge und seinen drei alternativ gespleißten Varianten in Gehirnregionen der Parkinson-Krankheit und in einem transgenen Mausmodell der Alpha-Synuclein-Überexpression. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternative Α-Synuclein-Transkriptverwendung als konvergenter Mechanismus in der Pathologie der Parkinson-Krankheit. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Kartographie des alternativen Spleißens von Neurexin, kartiert durch Einzelmolekül-Long-Read-mRNA-Sequenzierung. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). Veränderte Expression der FMR1-Spleißvariantenlandschaft bei Prämutationsträgern. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). Die Verbindung zwischen dem SNCA-Gen und Parkinsonismus. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Alternative polyadenylation of mRNAs: 3′-untranslated region matters in gene expression. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar