- Introduction
- Matériel et méthodes
- Échantillons de l’étude
- Extractions d’ADN génomique et d’ARN
- Préparation de la bibliothèque et séquençage
- Capture d’ADNg à l’aide de sondes IDT Xgen® Lockdown® et séquençage de molécules uniques
- Capture d’ADN à l’aide de sondes IDT Xgen® Lockdown® et séquençage d’isoformes à une seule molécule (Iso-Seq)
- Analyse de l’ADNg
- Analyse des variants courts et mise en phase
- Clustering and Determining Haplotypes for CT-Rich Region
- Analyse des isoformes
- Isoform SNP Calling
- Résultats
- La capture ciblée d’ADNg a permis d’identifier des variations connues et nouvelles
- La capture ciblée d’ADNc a identifié de nouveaux sites de début et de fin
- L’ADNc pleine longueur permet d’obtenir des informations de phasage au niveau de l’isoforme
- Discussion
- Data Availability
- Contributions des auteurs
- Financement
- Déclaration de conflit d’intérêts
- Remerciements
- Matériel supplémentaire
Introduction
Les programmes transcriptionnels et posttranscriptionnels contrôlent les niveaux d’expression des gènes et/ou la production de multiples isoformes d’ARNm distinctes, et les changements dans ces mécanismes entraînent une dérégulation de l’expression des gènes et des profils d’expression différentiels. Les régulations transcriptionnelles et posttranscriptionnelles aberrantes des gènes sont abondantes dans les tissus du système nerveux humain et contribuent aux différences phénotypiques au sein d’un même individu et entre individus, que ce soit dans un contexte de santé ou de maladie.
La dérégulation de l’expression de l’alpha-synucléine a été impliquée dans la pathogenèse des synucléinopathies, en particulier la maladie de Parkinson (PD) et la démence à corps de Lewy et (DLB). Alors que le rôle de la surexpression de la SNCA dans les synucléinopathies, principalement la MP, a été bien établi, nous nous sommes concentrés ici sur la détermination du répertoire complet des isoformes de transcription de la SNCA dans différentes synucléinopathies. Auparavant, plusieurs isoformes différentes du transcrit SNCA ont été décrites pour le gène SNCA, provenant de l’épissage alternatif, des sites de début de transcription (SCT) et de la sélection des sites de polyadénylation (McLean et al., 2012 ; Xu et al., 2014). L’épissage alternatif des exons codants donne naissance à SNCA 140, SNCA 112, SNCA 126 et SNCA 98, donnant lieu à quatre isoformes protéiques (Beyer et Ariza, 2012). Les SST alternatives du gène SNCA donnent lieu à quatre 5′UTR différentes, et la sélection alternative de différents sites de polyadénylation détermine trois longueurs majeures de la 3′UTR, sans impact sur la composition du produit protéique (Beyer et Ariza, 2012). Notre objectif global est d’acquérir de nouvelles connaissances sur la contribution des différentes espèces d’ARNm de la SNCA, connues et nouvelles, à la pathogenèse et à l’hétérogénéité des synucléinopathies.
À ce jour, la plupart des études ont utilisé des technologies de séquençage à lecture courte pour interroger la complexité du transcriptome dans les cerveaux humains. La disponibilité des technologies de troisième génération à lecture longue fournit une image sans précédent et presque complète des structures isoformes. Cependant, le séquençage existant des transcriptions à longue lecture pour les gènes de maladies humaines a utilisé une approche basée sur les amplicons (Treutlein et al., 2014 ; Kohli, 2017 ; Tseng et al., 2017). Bien que cette approche ait permis d’identifier un épissage alternatif complexe dans les gènes de maladies humaines, elle est limitée à la conception de l’amorce PCR et ne permet pas de découvrir les sites alternatifs de début et de fin. L’enrichissement ciblé, tel que l’utilisation de sondes IDT, peut fournir une vue complète des isoformes des gènes d’intérêt à un faible coût de séquençage. De plus, des lectures de transcription complète très précises permettent un haplotypage spécifique à l’isoforme.
Nous présentons ici la première étude connue utilisant la capture ciblée de l’ADNg et de l’ADNc de la région du gène SNCA à l’aide du séquençage PacBio SMRT. La région du gène SNCA est longue de ~114 kb, composée de six exons avec des longueurs de transcription d’environ 3 kb. Nous avons multiplexé 12 échantillons de cerveau humain provenant de la MP, de la DLB et d’échantillons de contrôle normaux et séquencé la bibliothèque d’ADNg et d’ADNc sur le système PacBio Sequel. Nous décrivons les analyses bioinformatiques utilisées pour identifier les SNP, les indels et les répétitions en tandem courtes pour la capture d’ADNg, et l’haplotypage au niveau des isoformes pour les données d’ADNc. Nous montrons que la capture ciblée est un moyen rentable d’étudier conjointement la variation génomique et l’épissage alternatif dans un gène neural lié à une maladie.
Matériel et méthodes
Échantillons de l’étude
La cohorte d’étude (N = 12) était composée d’individus présentant trois diagnostics neuropathologiques confirmés par autopsie : (1) MP (N = 4) ; (2) DLB (N = 4) ; et (3) des sujets cliniquement et neuropathologiquement normaux (N = 4). Les tissus cérébraux du cortex frontal ont été obtenus par l’intermédiaire de la Kathleen Price Bryan Brain Bank (KPBBB) de l’Université Duke, du programme de don de cerveau et de corps du Banner Sun Health Research Institute (Beach et al., 2015) et du Layton Aging and Alzheimer’s Disease Center de l’Oregon Health and Science University. Les phénotypes neuropathologiques ont été déterminés lors de l’examen post-mortem selon des méthodes standard bien établies suivant la méthode et les recommandations de pratique clinique de McKeith et ses collègues (McKeith et al., 1999, 2005). La densité de la pathologie LB (dans un ensemble standard de régions cérébrales) a reçu des scores de léger, modéré, sévère et très sévère. Les échantillons étudiés dans chaque groupe de diagnostic, PD et DLB, ont été soigneusement sélectionnés de manière à ce que la gravité des phénotypes clinicopathologiques soit similaire dans chaque pathologie. Tous les cerveaux présentaient des corps de Lewy (LB) dans le tronc cérébral, le limbe et le néocortex, tandis que la MP présentait des scores de McKeith sévères à très sévères dans le sous-nigra et l’amygdale. Tous les cerveaux n’indiquent pas de MA selon les critères CERAD et le stade de Braak et Braak = II. Les échantillons de cerveau neurologiquement sain ont été obtenus à partir de tissus post-mortem de sujets cliniquement normaux qui ont été examinés, dans la plupart des cas, dans l’année qui a suivi le décès et qui n’ont pas présenté de troubles cognitifs ou de parkinsonisme et des résultats neuropathologiques insuffisants pour diagnostiquer la MP, la maladie d’Alzheimer (MA) ou d’autres troubles neurodégénératifs. Tous les échantillons étaient de race blanche. Les données démographiques et neuropathologiques de ces sujets sont résumées dans le tableau supplémentaire 1. Le projet a été approuvé par le Duke Institution Review Board (IRB) qui a fourni une approbation éthique. Les méthodes ont été réalisées conformément aux directives et règlements pertinents.
Extractions d’ADN génomique et d’ARN
L’ADN génomique a été extrait des tissus cérébraux selon le protocole standard de Qiagen (Qiagen, Valencia, CA). L’ARN total a été extrait des échantillons de cerveau (100 mg) en utilisant le réactif TRIzol (Invitrogen, Carlsbad, CA), suivi d’une purification avec un kit RNeasy (Qiagen, Valencia, CA), selon le protocole du fabricant. La concentration d’ADNg et d’ARN a été déterminée par spectrophotométrie, et la qualité des échantillons d’ARN et l’absence de dégradation significative ont été confirmées par des mesures du numéro d’intégrité de l’ARN (RIN, tableau supplémentaire 1) en utilisant un bioanalyseur Agilent.
Préparation de la bibliothèque et séquençage
Capture d’ADNg à l’aide de sondes IDT Xgen® Lockdown® et séquençage de molécules uniques
Approximativement 2 μg de chaque échantillon d’ADNg ont été cisaillés à 6 kb à l’aide du Covaris g-TUBE et ligaturés avec des adaptateurs à code-barres. Un pool équimolaire de bibliothèque d’ADNg à code-barres 12-plex (2 μg au total) a été introduit dans la capture basée sur la sonde avec un panel de gènes SNCA conçu sur mesure.
Une bibliothèque SMRTBell a été construite en utilisant 626 ng d’ADNg capturé et réamplifié1.
Capture d’ADN à l’aide de sondes IDT Xgen® Lockdown® et séquençage d’isoformes à une seule molécule (Iso-Seq)
Environ 100-150 ng d’ARN total par réaction ont été transcrits de manière inverse à l’aide du kit de synthèse d’ADNc SMARTer de Clontech et de 12 oligo dT à code-barres spécifiques à l’échantillon (avec des séquences de code-barres PacBio 16mer, voir Méthodes supplémentaires). Trois réactions de transcription inverse (RT) ont été traitées en parallèle pour chaque échantillon. L’optimisation de la PCR a été utilisée pour déterminer le nombre optimal de cycles d’amplification pour les réactions PCR à grande échelle en aval. Une seule amorce (l’amorce IIA du kit Clontech SMARTer 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) a été utilisée pour toutes les réactions PCR post-RT. Les produits de PCR à grande échelle ont été purifiés séparément avec des billes AMPure PB 1X, et le bioanalyseur a été utilisé pour le CQ. Un pool équimolaire de bibliothèque d’ADNc à code-barres 12-plex (1 μg au total) a été introduit dans la capture basée sur la sonde avec un panel de gènes SNCA conçu sur mesure.
Une bibliothèque SMRTBell a été construite en utilisant 874 ng d’ADNc capturé et réamplifié2. Une cellule SMRT 1M (film de 6 h) a été séquencée sur la plateforme PacBio Sequel en utilisant la chimie 2.0.
Analyse de l’ADNg
Le séquençage des données d’ADNg à code-barres a été effectué sur trois cellules SMRT 1M en utilisant la chimie 2.0. Les données ont été démultiplexées en exécutant l’application Demultiplex Barcodes dans PacBio SMRT Link v6.0.
Analyse des variants courts et mise en phase
Des lectures de séquence circulaire consensuelle (CCS) ont été générées en utilisant SMRT Analysis 6.0 à partir de chaque ensemble de données démultiplexées et alignées sur le génome de référence hg19 en utilisant minimap2. Les doublons PCR provenant de l’amplification post-capture ont été identifiés en cartographiant les points d’extrémité et marqués à l’aide d’un script personnalisé. Les variants courts ont été appelés à l’aide de GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Après un premier passage de filtrage utilisant la profondeur de couverture et les métriques de qualité, les variants ont été inspectés manuellement dans IGV3. Si les variants n’étaient pas en phase avec les SNP voisins, ils ont été filtrés manuellement. Les sites de variants qui ont passé la curation manuelle ont été utilisés conjointement avec les alignements CCS dédupliqués pour la mise en phase par lecture avec WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Des sous-séquences alignées sur chr4 : 90742331-90742559 (hg19) ont été extraites pour chaque échantillon. Après avoir inspecté la distribution de taille de ces sous-séquences, elles ont été regroupées par taille et similarité de séquence en utilisant une combinaison de python et MUSCLE (Edgar, 2004), et une séquence consensus a été générée indépendamment pour chaque cluster.
Scripts et flux de travail personnalisés décrits plus en détail dans https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Analyse des isoformes
Le séquençage des données d’ADNc à code-barres a été effectué sur un SMRT Cell 1M sur le système PacBio Sequel en utilisant la chimie 2.0. L’analyse bioinformatique a été effectuée à l’aide de l’application IsoSeq3 dans l’analyse PacBio SMRT v6.0.0 pour obtenir des séquences d’isoformes de haute qualité et de pleine longueur (voir Méthodes supplémentaires pour plus d’informations).
Isoform SNP Calling
Les lectures de pleine longueur associées aux 41 isoformes finales des 12 échantillons ont été alignées sur le génome hg19 pour créer un pileup. Les bases ayant un VQ inférieur à 13 ont été exclues. Ensuite, à chaque position avec une couverture d’au moins 40 bases, un test exact de Fisher avec correction de Bonferroni a été appliqué avec un seuil de p de 0,01. Seuls les SNP de substitution qui ne sont pas proches de régions homopolymères (tronçons de 4 ou plus du même nucléotide) ont été appelés. Après l’appel des SNP, le génotype de chaque échantillon a été déterminé en comptant le nombre de lectures pleine longueur (FL) spécifiques à l’échantillon. Si un échantillon avait 5+ lectures FL soutenant à la fois la base de référence et la base alternative, il était hétérozygote. Si un échantillon avait 5+ lectures FL soutenant un allèle et moins de 5 lectures FL pour l’autre, il était homozygote. Sinon, il était non concluant. Les scripts sont disponibles à : https://github.com/Magdoll/cDNA_Cupcake.
Résultats
Nous avons conçu des sondes personnalisées pour le gène SNCA et effectué une capture ciblée à la fois de l’ADNg et de l’ADNc sur une bibliothèque multiplexée composée de 12 échantillons de cerveau humain provenant de PD, DLB et de contrôles normaux (figure 1, tableau supplémentaire 1). Les bibliothèques d’ADNg et d’ADNc ont été séquencées sur la plateforme PacBio Sequel. L’analyse bioinformatique a été réalisée à l’aide du logiciel PacBio, suivie d’une analyse personnalisée.
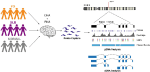
Figure 1. Présentation schématique de la conception de l’étude. Des matériaux d’ADN et d’ARN ont été extraits de tissus cérébraux post-mortem de patients atteints de la maladie de Parkinson, de la démence à corps de Lewy et de groupes témoins. Des bibliothèques d’ADNg et d’ADNc ont été constituées par hybridation de sonde et séquencées sur le système PacBio Sequel. L’analyse a été réalisée à l’aide du logiciel PacBio et d’autres outils existants.
La capture ciblée d’ADNg a permis d’identifier des variations connues et nouvelles
Après avoir généré des séquences consensus circulaires (CCS) et supprimé les doublons PCR (Méthodes supplémentaires), nous avons obtenu une couverture unique moyenne de 16 à 71 fois de la région du gène SNCA. Les lectures CCS avaient une longueur d’insertion moyenne de 2,9 kb et une précision de lecture moyenne de 98,9 %. À l’exception d’une région de 5 kb intentionnellement non couverte par les sondes en raison de la présence d’éléments LINE (hg19 chr4 : 90697216-90702113) et d’une région de 2,1 kb à forte teneur en GC autour de l’exon 1, la couverture était suffisante pour génotyper les deux haplotypes pour chacun des 212 échantillons (figure 2, figure supplémentaire 1).

Figure 2. Capture ciblée de l’ADNg et mise en phase. Un exemple montrant un échantillon de chaque condition. La piste supérieure montre l’une des isoformes de SNCA, suivie de la couverture de l’ADNg pour les trois échantillons. La piste des variantes montre chaque SNP et est codée en couleur pour les hétérozygotes (violet), les homozygotes alternatifs (orange) et les homozygotes de référence (gris). Les blocs en phase sont représentés en bleu clair. La piste du bas montre les emplacements des sondes de capture. La région d’abandon dans la conception de la sonde est due à deux éléments LINE au milieu de l’intron 4. Pour la couverture de l’ADNg et les informations de phasage des 12 échantillons, voir les figures supplémentaires.
En utilisant GATK4 HC, le filtrage basé sur la qualité et la curation manuelle, nous avons identifié 282 SNP et 35 indels, dont 8 SNPS et 13 indels non trouvés dans dbSNP (dbSNP Build ID : human_9606_b150_GRCh37p13) (tableau supplémentaire 2). Aucun variant n’a été identifié dans la région codante de SNCA, bien que huit variants aient été identifiés dans les régions non traduites. La majorité des variants identifiés, dont plusieurs répétitions en tandem courtes (STR), se situent dans les introns 2, 3 et 4.
Nous avons précédemment décrit une région riche en CT hautement polymorphe dans l’intron 4 de SNCA avec quatre haplotypes observés (Lutz et al., 2015). Bien que cette région hautement répétitive et structurellement variable se soit avérée difficile à génotyper avec GATK4 HC, nous avons pu construire des séquences consensus pour les 12 échantillons et avons observé les 4 haplotypes précédemment découverts (figure supplémentaire 2). De plus, nous avons identifié une nouvelle STR dans l’intron 4, consistant en une unité de trois bases répétée 16 fois dans la référence. Dans les 12 échantillons, nous avons identifié trois haplotypes, avec 9, 12 et 15 copies de l’unité répétée TTG. GATK HC a correctement génotypé tous ces haplotypes, à l’exception d’un haplotype pour PD-4, dont la couverture était assez faible dans cette région. Cependant, avec les données données pour cet échantillon, le génotype peut être déterminé par inspection visuelle (tableau 1).

Tableau 1. Un nouveau triplet de répétitions en tandem dans l’intron 4 (chr4 : 90713442).
Nous avons utilisé les variants courts détectés par GATK HC en conjonction avec l’outil de mise en phase basé sur les lectures WhatsHap (Martin et al., 2016) pour mettre en phase les lectures CCS à travers le locus, avec une gamme de succès dirigée principalement par la densité de variants hétérozygotes sur le locus. Les échantillons PD-1, PD-4, N-4, DLB-1 et DLB-4 présentaient de longues étendues de faible hétérozygotie, avec très peu de blocs de phase courts, tandis que les autres échantillons ont produit des blocs de phase allant de 7 à 18 fois la longueur de lecture moyenne, jusqu’à 54 kb (figure supplémentaire 3).
La capture ciblée d’ADNc a identifié de nouveaux sites de début et de fin
Nous avons traité les données d’ADNc PacBio (Iso-Seq) à l’aide du logiciel d’analyse PacBio SMRT. Après avoir mappé les données Iso-Seq sur hg19 et supprimé les artefacts (tableau supplémentaire 3, figure supplémentaire 4), nous avons obtenu un ensemble final de 41 isoformes SNCA (figure 3). Toutes les isoformes finales ont tous les sites d’épissage canoniques (GT-AG ou GC-AG) et sont soutenues par un total de 20 lectures de pleine longueur ou plus. La majorité des isoformes (28 sur 41) possèdent les six exons, ne différant que par l’utilisation de sites de départ 5′ alternatifs et de longueurs de 3′ UTR. Les longueurs de 3′ UTR variaient entre 300 et 2,6 kb. L’utilisation d’un site de départ 5′ alternatif très diversifié dans SNCA est connue ; ce qui l’est moins, c’est la longueur variable du 3′ UTR, qui avait été étudiée précédemment à l’aide de données RNA-seq qui ne permettaient pas de résoudre les structures d’isoformes de pleine longueur (Rhinn et al., 2012). Les données Iso-Seq montrent que la longueur 3′ UTR variable semble appariée à toutes les combinaisons possibles de sites de départ 5′, sans couplage préférentiel. Presque aucune des variabilités des sites de départ et d’arrivée ne modifie le cadre de lecture ouvert prédit (figure supplémentaire 5) et il est prévu qu’il se traduise par la séquence canonique de 141 acides aminés.

Figure 3. Les isoformes de SNCA capturées à l’aide de l’Iso-Seq ciblé identifient de nouveaux sites de début et de fin. La majorité de la complexité des isoformes provient de l’utilisation combinatoire de longueurs alternées de 3′ UTR et de l’exon 1, avec quelques rares sites d’épissage alternatifs trouvés dans l’exon 1 (vert), 2 (rouge) et 4 (bleu). Toutes les jonctions ont des sites d’épissage canoniques. Nous avons identifié cinq isoformes qui sautent l’exon 5 et deux isoformes qui sautent l’exon 3. Nous avons également identifié de nouveaux sites de début (orange) et de fin (violet) dans l’intron 4. Les SNP appelés sont marqués en violet.
Nous avons en outre validé les nouvelles jonctions (mais canoniques) en utilisant des données de jonction de lecture courte disponibles publiquement. La base de données Intropolis (v1, https://github.com/nellore/intropolis) combine plus de 21 000 ARN-seq disponibles publiquement. En raison du volume élevé de données de jonction soutenues par une seule lecture courte, pour cette étude, nous avons besoin d’un minimum de 10 lectures courtes (combinées à partir de tous les ensembles de données >21 000 RNA-seq) pour confirmer nos nouvelles jonctions Iso-Seq. À l’exception des nouvelles jonctions pour PB.1016.253 et PB.1016.296 (figure 3), toutes les autres nouvelles jonctions sont soutenues par l’ensemble de données Intropolis. Il est intéressant de noter que ces nouvelles jonctions sont beaucoup moins appuyées par les lectures courtes que les jonctions annotées par le Gencode. Par exemple, les deux nouvelles jonctions dans PB.1016.139 introduites par le nouvel exon sont soutenues par 2 519 et 44 lectures courtes Intropolis, respectivement, alors que les quatre autres jonctions connues sont soutenues par plus d’un million de lectures courtes. Cela montre la puissance de l’enrichissement ciblé utilisant le séquençage du transcriptome complet pour détecter des isoformes rares et nouvelles.
Nous avons observé deux isoformes avec saut d’exon 3 (SNCA126) et cinq isoformes avec saut d’exon 5 (SNCA112). Encore une fois, la diversité d’épissage dans ces deux groupes de saut d’exon provient principalement de l’utilisation diverse de sites de départ 5′ alternatifs et d’une longueur 3′ UTR variable. La prédiction de l’ORF montre que le saut de l’exon 3 ou de l’exon 5 raccourcit l’ORF mais maintient le cadre de lecture. Trois isoformes ont de nouveaux sites d’extrémité 3′ situés dans l’intron 4. La prédiction de l’ORF montre que cela entraîne un produit protéique tronqué.
Nous avons identifié un site de départ 5′ non annoté précédemment situé dans l’intron 4 (hg19 chr4 : 90692548-90693045, figure 3). Les trois isoformes associées à ce nouveau départ se composent du nouveau site de départ, de l’exon 5 et de longueurs 3′ UTR variables. Il est intéressant de noter que si les données de lecture courte téléchargées publiquement par GTEx et Sandor et al. (2017) et les données de pic CAGE (FANTOM5) n’appuient pas ce nouveau site de départ, un ensemble de données publiques récentes sur l’ARN direct NA128784 contenait un seul transcrit SNCA qui confirmait ce site de départ alternatif. En outre, la nouvelle jonction entre l’exon 5 et le nouveau site de départ est confirmée par les données de jonction de lecture courte d’Intropolis. Fait intéressant, ce nouveau site de départ 5′ est prédit pour introduire de nouveaux peptides tout en maintenant le cadre de lecture dans l’exon 5.
Nous avons également identifié trois transcriptions SNCA avec de nouveaux sites d’extrémité (Figure 3). Deux isoformes (PB.1016.383, PB.1016.384) utilisaient un 3′ UTR étendu dans l’exon 4, tandis que la troisième isoforme (PB.1016.381) utilisait un nouvel exon 3′ dans l’intron 4. Les nouvelles jonctions entre le dernier exon nouveau et l’exon précédent sont soutenues par les données publiques de jonction de lecture courte (Intropolis). Les nouveaux 3′ UTR donnent lieu à une prédiction d’ORF tronqué.
En utilisant le nombre de lectures normalisées sur toute la longueur comme indicateur de l’abondance des isoformes, nous constatons que l’une des isoformes canoniques de la SNCA (PB.1016.131) est la plus abondante, avec une abondance de 50-60 % dans tous les échantillons de sujets (tableau supplémentaire 4). Nous avons ensuite regroupé les 41 isoformes en fonction de leurs schémas d’épissage (tableau 2). Les isoformes qui possèdent les six exons représentent 95 à 97 % de l’abondance. Des études antérieures ont montré une augmentation marquée de l’expression des isoformes manquant l’exon 3 (SNCA126) dans le cortex frontal d’échantillons de DLB par rapport à la normale (Beyer et al., 2008) ; nos comptages d’isoformes agrégés montrent que trois des échantillons de DLB ont un niveau de comptage légèrement élevé par rapport aux échantillons normaux ainsi que les variantes SNCA112 (saut d’exon 5) pour la PD et la DLB par rapport aux échantillons normaux.

Tableau 2. Abondance de l’isoforme SNCA pour chaque échantillon, agrégée par les modèles d’épissage.
L’ADNc pleine longueur permet d’obtenir des informations de phasage au niveau de l’isoforme
Nous avons appelé les SNP à l’aide de l’ADNc en empilant toutes les lectures pleine longueur des 12 échantillons pour appeler les variants (voir la section “Méthodes”). Au total, quatre SNP ont été appelés et tous étaient précédemment annotés dans dbSNP (tableau 3, figure 3). Les quatre SNP sont tous situés dans des régions non-CDS, un dans le 3′ UTR (exon 6), un dans l’intron 4, et deux dans le 5′ UTR (exon 1). Le SNP 3′ UTR (chr4 : 90646886) est seulement couvert par les isoformes avec un 3′ UTR qui est au moins ~1 kb de long, et donc, toutes les isoformes canoniques ne couvrent pas ce SNP. Le SNP de l’intron 4 (chr4 : 90743331) est seulement couvert par les nouvelles isoformes alternatives de l’extrémité 3′ (PB.1016.383, PB.1016.384) et n’est relié à aucun des autres SNP. Les deux SNP 5′ UTR (chr4 : 90757312 et chr4 : 90758389) sont couverts par deux usages mutuellement exclusifs de l’exon 1 et ne sont donc pas non plus liés.

Tableau 3. Informations sur les SNP de l’ADNc.
Notre approche actuelle se limite à appeler uniquement des variantes de substitution dans les régions transcrites avec une couverture suffisante. La comparaison de la liste de nos SNP avec l’annotation hg19 dbSNP montre que la plupart des SNP ou variants manqués étaient soit d’une fréquence inférieure à 1% dans la population, soit n’étaient pas des substitutions de nucléotides simples, soit adjacents à des régions de faible complexité. Par exemple, le rs77964369 (chr4 : 90646532) est signalé comme ayant une fréquence de 50/50 de T/A ; cependant, ce T est adjacent à un tronçon de 11 As génomiques en aval. L’inspection manuelle de la pile de lectures Iso-Seq, qui compte ~1 300 lectures à cet endroit, ne suggère pas de preuve de variation, du moins parmi nos 12 échantillons.
À l’aide des lectures spécifiques à l’échantillon, nous appelons le génotype de chaque échantillon à chaque emplacement de SNP (tableau 3). En dehors de PD-2 qui a trop peu de lectures et qui n’est pas concluant pour les quatre SNP, nous avons pu appeler le génotype pour la plupart des autres échantillons. Notamment, DLB-3 est le seul échantillon qui est hétérozygote à tous les emplacements SNP. Sinon, nous n’avons pas observé de modèle spécifique à la condition de préférer un génotype à l’autre.
Discussion
Nous décrivons la première étude utilisant l’enrichissement ciblé du gène SNCA sur des bibliothèques multiplexées d’ADNg et d’ADNc pour étudier les maladies neurologiques en utilisant le séquençage à longue lecture. Les longues longueurs de lecture du système PacBio Sequel ont facilité le séquençage du répertoire complet des isoformes de transcription du gène SNCA. Nous avons révélé la diversité dans l’utilisation de sites de départ 5′ alternatifs et de longueurs 3′ UTR variables et observé des événements de saut d’exon connus, tels que la délétion de l’exon 3 (SNCA126) et la délétion de l’exon 5 (SNCA112). De plus, de nouveaux sites alternatifs de début et de fin dans le grand intron 4 ont été identifiés et il est prévu qu’ils soient traduits en nouvelles protéines. Il est probable que la grande profondeur de la couverture de séquençage de la capture ciblée, combinée à la capacité de séquencer des transcriptions complètes, nous a permis de détecter ces isoformes précédemment non décrites.
La signification biologique et pathologique des différentes isoformes de la protéine SNCA n’a pas encore été entièrement découverte. Cependant, des isoformes spécifiques de modification post-traductionnelle et d’épissage de la SNCA ont été associées à des propensions d’agrégation intracellulaire (Kalivendi et al., 2010) et sont exprimées différemment dans les synucléinopathies humaines (Beyer et al., 2008 ; Beyer et Ariza, 2012). Des études sur la modification post-traductionnelle du SNCA ont montré que les corps de Lewy, la marque pathologique des synucléinopathies, contiennent du SNCA phosphorylé, nitré et monoubiquitiné en abondance (Kim et al., 2014). Les effets des modifications post-transcriptionnelles sur l’agrégation de la SNCA ont également été étudiés. L’épissage alternatif a été suggéré pour affecter l’agrégation de la SNCA. La délétion de l’exon 3 ou 5 a des conséquences fonctionnelles : la délétion de l’exon 3 (SNCA126) entraîne l’interruption du domaine d’interaction protéine-membrane N-terminal, ce qui peut réduire l’agrégation, et la délétion de l’exon 5 (SNCA112) peut entraîner une agrégation accrue en raison d’un raccourcissement important de l’extrémité C-terminale non structurée (Lee et al., 2001 ; Beyer, 2006). Dans le cortex frontal des patients atteints de DLB, le taux de SNCA112 est nettement supérieur à celui des témoins (Beyer et al., 2008), tandis que les niveaux de SNCA126 sont réduits dans le cortex préfrontal des patients atteints de DLB (Beyer et al., 2006). En revanche, l’expression de SNCA126 a montré une augmentation dans le cortex frontal des cerveaux de personnes atteintes de la maladie de Parkinson et aucune différence significative dans l’ASM (Beyer et al., 2008). SNCA98 est une variante d’épissage spécifique au cerveau, dépourvue des exon 3 et 5, qui présente des niveaux d’expression différents dans diverses zones du cerveau fœtal et adulte. Une surexpression de SNCA98 a été rapportée dans les cortex frontaux de la DLB, de la PD (Beyer et al., 2007) et du MSA (Beyer et al., 2008) par rapport aux contrôles. En outre, le processus post transcriptionnel résultant de l’utilisation alternative de la 3′UTR a été signalé comme ayant des effets sur la stabilité et la localisation de l’ARNm (Fabian et al., 2010 ; Rhinn et al., 2012 ; Yeh et Yong, 2016). Des recherches supplémentaires concernant les propensions à l’agrégation des différentes isoformes connues de la protéine SNCA et la composition des corps de Lewy sont justifiées. En outre, notre étude a jeté les bases d’analyses de quantification de l’ARNm des transcriptions connues et nouvelles dans un échantillon plus important composé de sujets présentant différents stades clinicopathologiques et utilisant plusieurs régions du cerveau de chaque sujet. Ces analyses du paysage transcriptomique de la SNCA spécifique à la région cérébrale dans le contexte de la gravité neuropathologique seront informatives en ce qui concerne le rôle des isoformes spécifiques de la SNCA dans la progression des stades neuropathologiques et la gravité de la densité des corps de Lewy et des neurites de Lewy.
Dans cet article, nous nous sommes concentrés sur la création d’une norme de séquençage et d’analyse pour analyser les données ciblées d’ADNg et d’ADNc générées à partir des mêmes sujets. Il s’agit d’une approche puissante qui permet potentiellement la mise en phase des séquences d’ADNg à travers la région complète d’un gène particulier en fonction de l’hétérozygotie dans la séquence des isoformes de transcription pleine longueur. Les données d’ADNg ciblées par PacBio dans cette étude ont produit des blocs phasés qui couvrent 81% de la région de 114 kb centrée sur SNCA, le bloc phasé le plus long dépassant 54 kb. Comme la mise en phase de l’ADNg est limitée par la longueur de lecture et l’hétérozygotie, l’augmentation de la longueur de lecture générera probablement des blocs de phase plus grands.
L’analyse des variantes de l’ADNg a confirmé les répétitions en tandem courtes (STR) connues et identifié de nouvelles dans les régions introniques. Par exemple, précédemment, en utilisant le séquençage en phase par clonage et le séquençage Sanger, nous avons découvert quatre haplotypes distincts dans une région intronique riche en CT qui comprenait un groupe de séquences répétitives variables (Lutz et al., 2015). Nous avons montré qu’un haplotype spécifique, appelé haplotype 3, conférait un risque de développer une pathologie à corps de Lewy chez les patients atteints de la maladie d’Alzheimer. Ici, nous avons validé la séquence de cette région hautement polymorphe de faible complexité et ses quatre haplotypes définis. Bien que la taille de notre échantillon soit faible, l'” haplotype 3 ” était présent exclusivement chez les patients atteints de la maladie (un patient PD, deux patients DLB), ce qui correspond à nos résultats précédents. Les résultats pilotes et notre publication précédente fournissent la prémisse pour répéter les analyses d’association des synucléinopathies avec des STRs et des haplotypes structurels définis avec précision, c’est-à-dire par de longues lectures, en utilisant une taille d’échantillon plus importante.
Notre article a démontré la capacité du système PacBio Sequel à découvrir de nouveaux transcrits complets et à caractériser le répertoire complet des transcrits complets d’un gène impliqué dans une maladie. De plus, nous avons également montré que les lectures longues de l’ADNg définissent avec plus de précision les variants structurels courts et les haplotypes, y compris les STR, ce qui peut faciliter la découverte et la validation de variants associés à la maladie autres que les SNP. Collectivement, ces nouvelles connaissances sont très précieuses et applicables pour faire progresser notre compréhension des étiologies génétiques, qui peuvent impliquer des perturbations dans le paysage de transcription, sous-jacentes aux maladies humaines complexes, y compris les troubles neurodégénératifs liés à l’âge tels que les synucléinopathies.
Data Availability
Les trois cellules SMRT de données brutes d’ADNg sont disponibles sur Zenodo.org avec doi : 10.5281/zenodo.1560688. La cellule SMRT des données brutes d’ADNc est disponible sur Zenodo.org avec le doi : 10.5281/zenodo.1581809. Les résultats traités d’ADNg et d’ADNc, y compris les variantes d’ADNg et les isoformes d’ADNc, sont disponibles à Zenodo.org avec doi : 10.5281/zenodo.3261805.
Contributions des auteurs
OC-F a contribué à la conception et au design de l’étude. ET et WR ont organisé les bases de données de séquences, effectué les analyses de séquençage et préparé toutes les figures et tous les tableaux. O-CG et JB ont manipulé les tissus cérébraux et les préparations d’échantillons nucléiques. TH a généré les ensembles de données de séquençage. SK a conçu et obtenu les réactifs. OC-F, ET et WR ont rédigé la première version du manuscrit. OC-F a obtenu un financement. Tous les auteurs ont contribué à la préparation du manuscrit, lu et approuvé la version soumise.
Financement
Ce travail a été financé en partie par les National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Déclaration de conflit d’intérêts
ET, WR, TH et SK sont ou étaient des employés de Pacific Biosciences au moment de l’étude.
Les autres auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière qui pourrait être interprétée comme un conflit d’intérêts potentiel.
Remerciements
Ce manuscrit a été publié en tant que préimpression à BioRxiv (Tseng et al, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Matériel supplémentaire
Le matériel supplémentaire pour cet article peut être trouvé en ligne à : https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathologie 35, 354-389. doi : 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). La structure de la Α-synucléine, la modification post-traductionnelle et l’épissage alternatif en tant qu’exhausteurs d’agrégation. Acta Neuropathol. 112, 237-251. doi : 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., et Ariza, A. (2012). La modification post-traductionnelle de l’alpha-synucléine et l’épissage alternatif comme déclencheur de la neurodégénérescence. Mol. Neurobiol. 47, 509-524. doi : 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., and Ariza, A. (2008). Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 9, 163-172. doi : 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Identification et caractérisation d’une nouvelle isoforme d’alpha-synucléine et son rôle dans les maladies à corps de Lewy. Neurogenetics 9, 15-23. doi : 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. Neuroreport 17, 1327-1330. doi : 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., et Filipowicz, W. (2010). Régulation de la traduction et de la stabilité des ARNm par les microARN. Annu. Rev. Biochem. 79, 351-379. doi : 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE : alignement de séquences multiples avec une grande précision et un débit élevé. Nucleic Acids Res. 32, 1792-1797. doi : 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Les oxydants induisent un épissage alternatif de la Α-synucléine : implications pour la maladie de Parkinson. Free Radic. Biol. Med. 48, 377-383. doi : 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., et Halliday, G. M. (2014). La biologie de l’alpha-synucléine dans les maladies à corps de Lewy. Alzheimers Res. Ther. 6, 1-9. doi : 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). La variante du récepteur des androgènes AR-V9 est coexprimée avec AR-V7 dans les métastases du cancer de la prostate et prédit la résistance à l’abiratérone. Clin. Cancer Res. 23, 1-13. doi : 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., et Lee, S. J. (2001). La Α-synucléine liée à la membrane a une forte propension à l’agrégation et la capacité d’ensemencer l’agrégation de la forme cytosolique. J. Biol. Chem. 277, 671-678. doi : 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., et Chiba-Falek, O. (2015). Un haplotype riche en cytosine-thymine (CT) dans l’intron 4 de SNCA confère un risque de pathologie à corps de Lewy dans la maladie d’Alzheimer et affecte l’expression de SNCA. Alzheimers Dement. 11, 1133-1143. doi : 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap : fast and accurate read-based phasing. bioRxiv . doi : 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnostic et gestion de la démence à corps de Lewy : troisième rapport du consortium DLB. Neurology 65, 1863-1872. doi : 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Rapport du deuxième atelier international sur la démence à corps de Lewy : diagnostic et traitement. Consortium sur la démence à corps de Lewy. Neurology 53, 902-905. doi : 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., et Isacson, O. (2012). Niveaux d’expression transcriptionnelle de l’alpha-synucléine pleine longueur et de ses trois variantes alternativement épissées dans les régions cérébrales de la maladie de Parkinson et dans un modèle de souris transgénique de surexpression de l’alpha-synucléine. Mol. Cell. Neurosci. 49, 230-239. doi : 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv . doi : 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). L’utilisation alternative des transcriptions de la Α-synucléine comme mécanisme convergent dans la pathologie de la maladie de Parkinson. Nat. Commun. 3, 889-821. doi : 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Le profilage transcriptomique des neurones dopaminergiques purifiés dérivés de patients identifie des perturbations convergentes et des thérapeutiques pour la maladie de Parkinson. Hum. Mol. Genet. 54, ddw412-ddw415. doi : 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., et Südhof, T. C. (2014). Cartographie de l’épissage alternatif de la neurexine cartographiée par le séquençage de l’ARNm à lecture longue à une seule molécule. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi : 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). Le paysage des transcrits SNCA à travers les synucléinopathies : de nouveaux aperçus à partir de l’analyse de séquençage à long lit. bioRxiv . doi : 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., et Tassone, F. (2017). L’expression altérée du paysage des variants d’épissage de FMR1 chez les porteurs de prémutation. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi : 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., et Yu, J.-T. (2014). Le lien entre le gène SNCA et le parkinsonisme. Neurobiol. Aging 36, 1-14. doi : 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., et Yong, J. (2016). La polyadénylation alternative des ARNm : La région 3′ non traduite compte dans l’expression des gènes. Mol. Cell 39, 281-285. doi : 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar
.