- Introduction
- Anyagok és módszerek
- Vizsgálati minták
- Genomi DNS és RNS kivonás
- Library Preparation and Sequencing
- gDNS befogása IDT Xgen® Lockdown® Probes és Single-Molecule Sequencing segítségével
- cDNS befogása IDT Xgen® Lockdown® szondák használatával és egymolekulás izoforma-szekvenálás (Iso-Seq)
- gDNS-elemzés
- Rövid variánselemzés és fáziskódolás
- Clustering and Determining Haplotypes for CT-Rich Region
- Isoformák elemzése
- Isoform SNP Calling
- Eredmények
- A célzott gDNS-felvétel ismert és új variációkat azonosított
- Célzott cDNS befogás azonosított új kezdő- és végpontokat
- A teljes hosszúságú cDNS lehetővé teszi az izoforma-szintű fázisinformációt
- Diszkusszió
- Adatok elérhetősége
- Author Contributions
- Finanszírozás
- Conflict of Interest Statement
- Megköszönések
- Kiegészítő anyag
Introduction
Transcriptional and posttranscriptional programs control gene expression levels and/or production of multiple distinct mRNS isoforms, and changes in these mechanisms result in dysregulation of gene expression and differential expression profiles. Az aberráns transzkripciós és poszttranszkripciós génszabályozás gyakori az emberi idegrendszer szöveteiben, és hozzájárul az egyéneken belüli és az egyének közötti fenotípusos különbségekhez egészségben és betegségben.
Az alfa-szinuklein expressziójának diszregulációja szerepet játszik a szinukleinopátiák, különösen a Parkinson-kór (PD) és a Lewy-testes demencia (DLB) patogenezisében. Míg az SNCA overexpressziójának szerepe a szinukleinopátiákban, főként a Parkinson-kórban, jól ismert, itt az SNCA transzkript izoformák teljes repertoárjának meghatározására összpontosítottunk a különböző szinukleinopátiákban. Korábban több különböző SNCA transzkript izoformát írtak le az SNCA génre vonatkozóan, amelyek alternatív splicing, transzkripciós starthelyek (TSS-ek) és a poliadenilációs helyek kiválasztása révén keletkeztek (McLean és mtsai., 2012; Xu és mtsai., 2014). A kódoló exonok alternatív splicingje az SNCA 140, SNCA 112, SNCA 126 és SNCA 98 géneket eredményezi, amelyek négy fehérjeizoformát eredményeznek (Beyer és Ariza, 2012). Az SNCA gén alternatív TSS-jei négy különböző 5′UTR-t eredményeznek, és a különböző poliadenilációs helyek alternatív kiválasztása a 3′UTR három fő hosszát határozza meg, amelyeknek nincs hatása a fehérjetermék összetételére (Beyer és Ariza, 2012). Átfogó célunk, hogy új betekintést nyerjünk a különböző – ismert és új – SNCA mRNS-fajok hozzájárulására a synucleinopathiák patogeneziséhez és heterogenitásához.
A mai napig a legtöbb tanulmány rövid olvasású szekvenálási technológiákat használt az emberi agyak transzkriptomjának komplexitásának vizsgálatára. A harmadik generációs hosszú olvasási technológiák elérhetősége példátlan és szinte teljes képet nyújt az izoformák szerkezetéről. A meglévő hosszú olvasású transzkriptum-szekvenálások azonban a humán betegségek génjeire vonatkozóan amplikonalapú megközelítést alkalmaztak (Treutlein és mtsai., 2014; Kohli, 2017; Tseng és mtsai., 2017). Bár ez a megközelítés sikeres volt a humán betegséggének komplex alternatív splicingjének azonosításában, a PCR-primerek tervezésére korlátozódik, és nem tárja fel az alternatív kezdő- és végpontokat. A célzott dúsítás, például IDT-szondák alkalmazásával, alacsony szekvenálási költségek mellett átfogó izoforma-áttekintést nyújthat az érdeklődésre számot tartó génekről. Továbbá a nagy pontosságú teljes hosszúságú transzkriptolvasatok lehetővé teszik az izoforma-specifikus haplotipizálást.
Itt mutatjuk be az első ismert tanulmányt, amely az SNCA gén régió gDNS-ének és cDNS-ének célzott befogását alkalmazza PacBio SMRT szekvenálással. Az SNCA gén régiója ~114 kb hosszú, hat exonból áll, amelyek transzkripthossza 3 kb körüli. 12 humán agymintát multiplexeltünk PD, DLB és normál kontroll mintákból, és a gDNS és cDNS könyvtárat szekvenáltuk a PacBio Sequel rendszeren. Leírjuk az SNP-k, indelek és rövid tandem ismétlődések azonosítására használt bioinformatikai elemzéseket a gDNS-felvételek esetében, valamint az izoforma-szintű haplotipizálást a cDNS-adatok esetében. Megmutatjuk, hogy a célzott rögzítés költséghatékony módja a genomi variáció és az alternatív splicing együttes vizsgálatának egy betegséggel kapcsolatos neurális génben.
Anyagok és módszerek
Vizsgálati minták
A vizsgálati kohorsz (N = 12) három boncolással megerősített neuropathológiai diagnózissal rendelkező személyekből állt: (1) Parkinson-kór (N = 4); (2) DLB (N = 4); és (3) klinikailag és neuropatológiailag normális személyek (N = 4). A frontális kéreg agyszöveteit a Duke Egyetem Kathleen Price Bryan Brain Bank (KPBBB), a Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) és az Oregon Health and Science University Layton Aging and Alzheimer’s Disease Center (Layton Öregedés és Alzheimer-kór Központ) segítségével nyerték. A neuropatológiai fenotípusokat postmortem vizsgálat során határozták meg a standard, jól bevált módszereket követve, a McKeith és munkatársai (McKeith et al., 1999, 2005) módszerét és klinikai gyakorlati ajánlásait követve (McKeith et al., 1999, 2005). Az LB-patológia sűrűsége (az agyi régiók standard készletében) enyhe, közepes, súlyos és nagyon súlyos pontszámokat kapott. A vizsgálati mintákat az egyes diagnóziscsoportokon belül – PD és DLB – gondosan úgy választottuk ki, hogy a klinikopatológiai fenotípusok súlyossága hasonló legyen az egyes patológiákon belül. Minden agytörzsben agytörzsi, limbikus és neokortikális Lewy-testeket (LB) mutattak ki, míg a PD-ben súlyos vagy nagyon súlyos McKeith-pontszámokat mutattak ki a subnigra és az amygdala területén. Valamennyi agy a CERAD-kritériumok és a Braak és Braak-stádium = II szerint nem jelez AD-t. A neurológiailag egészséges agymintákat olyan klinikailag normális alanyok postmortem szöveteiből nyerték, akiket a legtöbb esetben a halálukat követő 1 éven belül vizsgáltak meg, és akiknél nem találtak kognitív zavart vagy parkinsonizmust, és a neuropatológiai leletek nem elegendőek a Parkinson-kór, az Alzheimer-kór (AD) vagy más neurodegeneratív rendellenességek diagnosztizálásához. Minden minta fehér bőrű volt. Ezen alanyok demográfiai és neuropatológiai adatait az 1. kiegészítő táblázat foglalja össze. A projektet a Duke intézményi felülvizsgálati bizottsága (IRB) hagyta jóvá, amely etikai jóváhagyást adott. A módszereket a vonatkozó irányelveknek és előírásoknak megfelelően végeztük.
Genomi DNS és RNS kivonás
A genomi DNS kivonása agyszövetekből a Qiagen standard protokollja szerint történt (Qiagen, Valencia, CA). A teljes RNS-t agymintákból (100 mg) TRIzol reagenssel (Invitrogen, Carlsbad, CA) extraháltuk, majd a gyártó protokollját követve RNeasy kit (Qiagen, Valencia, CA) segítségével tisztítottuk. gDNS és RNS koncentrációját spektrofotometriásan határoztuk meg, az RNS minták minőségét és a jelentős degradáció hiányát pedig az RNS integritási szám (RIN, 1. kiegészítő táblázat) mérésével igazoltuk Agilent Bioanalyzer segítségével.
Library Preparation and Sequencing
gDNS befogása IDT Xgen® Lockdown® Probes és Single-Molecule Sequencing segítségével
Minden gDNS-mintából körülbelül 2 μg-ot 6 kb-ra nyírtunk a Covaris g-TUBE segítségével, és vonalkódolt adapterekkel ligáltuk. A 12-plex vonalkódolt gDNS-könyvtár egy ekvimoláris poolját (összesen 2 μg) vittük be a szonda alapú rögzítésbe egy egyedi tervezésű SNCA génpanellel.
A SMRTBell könyvtárat 626 ng rögzített és újraamplikált gDNS1 felhasználásával hoztuk létre.
cDNS befogása IDT Xgen® Lockdown® szondák használatával és egymolekulás izoforma-szekvenálás (Iso-Seq)
Reakciónként kb. 100-150 ng teljes RNS-t reverz transzkripcióztunk a Clontech SMARTer cDNS-szintézis kit és 12 mintaspecifikus vonalkódolt oligo dT (PacBio 16mer vonalkódszekvenciákkal, lásd Kiegészítő módszerek) segítségével. Minden minta esetében három reverz transzkripciós (RT) reakciót dolgoztunk fel párhuzamosan. PCR-optimalizálással határoztuk meg az optimális amplifikációs ciklusszámot a későbbi nagyméretű PCR-reakciókhoz. Egyetlen primert (a Clontech SMARTer kit IIA primere 5′ AAG CAG CAG TGG TAT CAA CGC AGA GTA C 3′) használtunk az összes RT utáni PCR-reakcióhoz. A nagy PCR-termékeket külön tisztítottuk 1X AMPure PB gyöngyökkel, és a bioanalizátort használtuk a minőségellenőrzéshez. Egy 12-plex vonalkódolt cDNS-könyvtár (összesen 1 μg) ekvimoláris poolját vittük be a szonda alapú rögzítésbe egy egyedi tervezésű SNCA génpanellel.
A SMRTBell könyvtárat 874 ng rögzített és újraamplikált cDNS2 felhasználásával állítottuk elő. Egy SMRT Cell 1M (6 h film) szekvenálását végeztük el a PacBio Sequel platformon 2.0 kémia használatával.
gDNS-elemzés
A vonalkódolt gDNS-adatok szekvenálását három SMRT Cell 1M-en végeztük el 2.0 kémia használatával. Az adatokat a PacBio SMRT Link v6.0-ban a Demultiplex Barcodes alkalmazás futtatásával demultiplexáltuk.
Rövid variánselemzés és fáziskódolás
A SMRT Analysis 6.0 segítségével minden demultiplexált adatsorból körkörös konszenzus szekvencia (CCS) olvasatokat generáltunk és minimap2 segítségével a hg19 referencia genomhoz igazítottuk. A befogás utáni amplifikációból származó PCR-duplikátumokat a végpontok feltérképezésével azonosítottuk, és egyedi szkript segítségével megjelöltük. A rövid variánsokat a GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018) segítségével hívtuk meg. A lefedettségi mélység és a minőségi metrikák segítségével végzett első szűrés után a variánsokat kézzel ellenőriztük az IGV3-ban. Ha a variánsok nem fázisoltak a közeli SNP-kkel, akkor kézzel szűrtük őket. A manuális kuráción átesett variánshelyeket a deduplikált CCS-illesztésekkel együtt használtuk a WhatsHap (Martin et al., 2016) segítségével történő olvasásalapú fázistoláshoz.
Clustering and Determining Haplotypes for CT-Rich Region
A chr4: 90742331-90742559 (hg19) chr4: 90742331-90742559 (hg19) szekvenciákhoz igazított alszekvenciákat minden mintához kivontuk. Miután megvizsgáltuk e részszekvenciák méreteloszlását, a python és a MUSCLE (Edgar, 2004) kombinációjának segítségével méret és szekvencia-hasonlóság alapján klasztereztük őket, és minden egyes klaszterhez függetlenül létrehoztunk egy konszenzusszekvenciát.
Az egyedi szkriptek és munkafolyamatok a továbbiakban a https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases pontban vannak leírva.
Isoformák elemzése
A vonalkódolt cDNS-adatok szekvenálása egy SMRT Cell 1M-en történt a PacBio Sequel rendszeren, 2.0 vegyszerrel. A bioinformatikai elemzést a PacBio SMRT Analysis v6.0.0.0 rendszer IsoSeq3 alkalmazásával végeztük, hogy kiváló minőségű, teljes hosszúságú izoforma szekvenciákat kapjunk (további információkért lásd a Kiegészítő módszerek című részt).
Isoform SNP Calling
A 12 minta végső 41 izoformájához kapcsolódó teljes hosszúságú olvasatokat a hg19 genomhoz igazítottuk, hogy egy pileupot hozzunk létre. A 13-nál kisebb QV-vel rendelkező bázisokat kizártuk. Ezután minden egyes, legalább 40 bázissal lefedett pozícióban Fisher-féle egzakt tesztet alkalmaztunk Bonferroni korrekcióval, 0,01-es p cutoff értékkel. Csak a homopolimer régiókhoz (4 vagy több azonos nukleotidból álló szakaszok) nem közeli szubsztitúciós SNP-ket neveztük meg. Az SNP-k meghívása után az egyes minták genotípusát a támogató mintaspecifikus teljes hosszúságú (FL) olvasatok számának összeszámlálásával határoztuk meg. Ha egy minta 5+ FL olvasattal rendelkezett, amely mind a referencia-, mind az alternatív bázist támogatta, akkor heterozigóta volt. Ha egy mintában 5+ FL olvasat támogatta az egyik allélt, és kevesebb mint 5 FL olvasat a másik allélt, akkor homozigóta volt. Ellenkező esetben nem volt egyértelmű. A szkriptek elérhetők a következő címen: https://github.com/Magdoll/cDNA_Cupcake.
Eredmények
Egyedi szondákat terveztünk az SNCA génhez, és mind a gDNS, mind a cDNS célzott befogását elvégeztük egy multiplexelt könyvtáron, amely 12 emberi agymintából állt, amelyek PD, DLB és normál kontrolloktól származnak (1. ábra, 1. kiegészítő táblázat). A gDNS- és cDNS-könyvtárakat a PacBio Sequel platformon szekvenáltuk. A bioinformatikai elemzést a PacBio szoftver segítségével végeztük, amelyet egyéni elemzés követett.
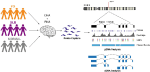
1. Ábra. A vizsgálati terv sematikus bemutatása. A DNS- és RNS-anyagokat Parkinson-kóros, Lewy-testes demenciában szenvedő betegek és kontrollcsoportok postmortem agyszöveteiből vontuk ki. gDNS- és cDNS-könyvtárakat készítettünk szondahibridizációval és szekvenáltuk a PacBio Sequel rendszeren. Az elemzést a PacBio szoftver és más meglévő eszközök segítségével végeztük.
A célzott gDNS-felvétel ismert és új variációkat azonosított
A cirkuláris konszenzus szekvenciák (CCS) előállítása és a PCR duplikátumok eltávolítása után (Kiegészítő módszerek) 16-71-szeres átlagos egyedi lefedettséget kaptunk az SNCA gén régiójában. A CCS leolvasások átlagos inszerthossza 2,9 kb, átlagos leolvasási pontossága pedig 98,9% volt. Egy 5 kb-os régió kivételével, amelyet a LINE elemek jelenléte miatt szándékosan nem fedtek le a szondák (hg19 chr4: 90697216-90702113) és egy 2,1 kb-os, magas GC-tartalmú régiót az 1. exon körül, elegendő lefedettség állt rendelkezésre mindkét haplotípus genotipizálására mind a 212 minta esetében (2. ábra, 1. kiegészítő ábra).

2. ábra. Célzott gDNS-befogás és fázistolás. Egy példa, amely minden feltételből egy-egy mintát mutat. A felső sáv az egyik SNCA izoformát mutatja, majd a három minta gDNS-lefedettségét. A variáns sáv az egyes SNP-ket mutatja, és színkódolással jelöli a heterozigóta (lila), a homozigóta alternatív (narancssárga) és a homozigóta referencia (szürke) SNP-ket. A fázisos blokkok világoskékkel vannak jelölve. Az alsó sáv a befogási szondák helyét mutatja. A szondatervezésben a kieső régiót a 4. intron közepén lévő két LINE elem okozza. Mind a 12 minta gDNS-lefedettségét és fázistérképét lásd a Kiegészítő ábrákon.
A GATK4 HC, a minőségalapú szűrés és a kézi kuráció segítségével 282 SNP-t és 35 indelt azonosítottunk, köztük 8 SNPS-t és 13 indelt, amelyek nem találhatók a dbSNP-ben (dbSNP Build ID: human_9606_b150_GRCh37p13) (Kiegészítő táblázat 2). Az SNCA kódoló régiójában nem azonosítottak variánsokat, bár nyolc variánst azonosítottak a nem transzlált régiókban. Az azonosított variánsok többsége, köztük számos rövid tandemismétlődés (STR), a 2., 3. és 4. intronba esik.
Az SNCA 4. intronjában korábban leírtunk egy erősen polimorf CT-gazdag régiót négy megfigyelt haplotípussal (Lutz és mtsai., 2015). Bár ezt az erősen repetitív és szerkezetileg változó régiót nehéznek bizonyult genotipizálni a GATK4 HC-vel, mind a 12 minta esetében konszenzus szekvenciákat tudtunk konstruálni, és mind a 4 korábban felfedezett haplotípust megfigyeltük (2. kiegészítő ábra). Ezenkívül azonosítottunk egy új STR-t a 4. intronban, amely a referenciában 16-szor megismételt három bázisegységből áll. A 12 mintán belül három haplotípust azonosítottunk, a TTG ismétlődő egység 9, 12 és 15 példányával. A GATK HC mindegyiket helyesen genotipizálta, kivéve a PD-4 egy haplotípusát, amely meglehetősen alacsony lefedettséggel rendelkezett ebben a régióban. A minta adott adatai alapján azonban a genotípus vizuálisan is meghatározható (1. táblázat).

Táblázat. Új triplet tandem ismétlődés a 4. intronban (chr4: 90713442).
A GATK HC által detektált rövid variánsokat a WhatsHap (Martin et al., 2016) olvasásalapú fáziskészítő eszközzel együtt használtuk a CCS olvasatok fáziskészítésére a lókuszon keresztül, a sikeresség tartományát leginkább a lókusz feletti heterozigóta változatsűrűség határozta meg. A PD-1, PD-4, N-4, DLB-1 és DLB-4 minták hosszú, alacsony heterozigozitású szakaszokat mutattak, nagyon kevés, rövid fázisblokkal, míg a többi minta az átlagos olvasási hossz 7-18-szorosától az 54 kb-ig terjedő fázisblokkokat eredményezett (3. kiegészítő ábra).
Célzott cDNS befogás azonosított új kezdő- és végpontokat
A PacBio cDNS (Iso-Seq) adatait a PacBio SMRT Analysis szoftverrel dolgoztuk fel. Az Iso-Seq-adatok hg19-re történő leképezése és az artefaktumok eltávolítása után (3. kiegészítő táblázat, 4. kiegészítő ábra) 41 SNCA izoformát tartalmazó végleges készletet kaptunk (3. ábra). Minden végleges izoforma rendelkezik az összes kanonikus splice-helyekkel (GT-AG vagy GC-AG), és összesen 20 vagy annál több teljes hosszúságú olvasat támasztja alá. Az izoformák többsége (41-ből 28) mind a hat exonnal rendelkezik, és csak az alternatív 5′ start helyek és a 3′ UTR hosszában különbözik. A 3′ UTR hossza 300 és 2,6 kb között változott. A rendkívül változatos alternatív 5′ starthely használata az SNCA-ban ismert; ami kevésbé ismert, az a változó 3′ UTR-hossz, amelyet korábban olyan RNS-seq adatok alapján vizsgáltak, amelyek nem oldották fel a teljes hosszúságú izoformák szerkezetét (Rhinn és mtsai., 2012). Az Iso-Seq adatok azt mutatják, hogy a változó 3′ UTR-hossz, úgy tűnik, az 5′ starthelyek minden lehetséges kombinációjával párosul, preferenciális párosítás nélkül. A kezdő- és véghely variabilitása szinte egyáltalán nem változtatja meg a prediktált nyílt olvasási keretet (5. kiegészítő ábra), és az előrejelzések szerint a 141 aminosavból álló kanonikus szekvenciára transzlálódik.

3. ábra. A célzott Iso-Seq segítségével rögzített SNCA izoformák új kezdő- és végpontokat azonosítanak. Az izoforma-komplexitás nagy része az alternatív 3′ UTR-hosszúságok és az 1. exon kombinatorikus használatából származik, néhány ritka alternatív splice-hely található az 1. (zöld), 2. (piros) és 4. (kék) exonban. Minden elágazás kanonikus splice-helyekkel rendelkezik. Öt olyan izoformát azonosítottunk, amely kihagyta az 5. exont, és két olyan izoformát, amely kihagyta a 3. exont. Új kezdő (narancssárga) és végpontokat (lila) is azonosítottunk a 4-es intronban. A kinevezett SNP-ket lilával jelöltük.
Az új (de kanonikus) kapcsolódási pontokat nyilvánosan elérhető rövid olvasású kapcsolódási adatok segítségével tovább validáltuk. Az Intropolis (v1, https://github.com/nellore/intropolis) adatbázis több mint 21 000 nyilvánosan elérhető RNS-seq adatot egyesít. A csak egyetlen rövid leolvasással támogatott csomóponti adatok nagy mennyisége miatt ehhez a vizsgálathoz legalább 10 rövid leolvasás támogatására van szükségünk (az összes >21 000 RNA-seq-adatkészletből kombinálva) az Iso-Seq új csomópontjaink megerősítéséhez. A PB.1016.253 és PB.1016.296 új csomópontok kivételével (3. ábra) az összes többi új csomópontot támogatja az Intropolis adathalmaz. Érdekes módon ezek az új csomópontok lényegesen kevesebb rövid olvasatot támogatnak, mint a Gencode-annotált csomópontok. Például a PB.1016.139-ben az új exon által bevezetett két új csomópontot 2519, illetve 44 Intropolis rövid olvasatszám támogatja, míg a másik négy ismert csomópontot több mint 1 millió rövid olvasatszám támogatja. Ez mutatja a teljes hosszúságú transzkriptom szekvenálással végzett célzott dúsítás erejét a ritka, új izoformák kimutatására.
Két izoformát figyeltünk meg 3. exon kihagyásával (SNCA126) és öt izoformát 5. exon kihagyásával (SNCA112). A splicing diverzitás ebben a két exon skipping csoportban ismét leginkább az alternatív 5′ start helyek változatos használatából és a változó 3′ UTR hosszból ered. Az ORF-előrejelzés azt mutatja, hogy a 3. vagy 5. exon kihagyása megrövidíti az ORF-et, de megtartja az olvasási keretet. Három izoformának a 4. intronban található új 3′ végpontja van. Az ORF-előrejelzés azt mutatja, hogy ez csonka fehérjeterméket eredményez.
A 4. intronban (hg19 chr4: 90692548-90693045, 3. ábra) azonosítottunk egy korábban nem jegyzett 5′ starthelyet. Az ehhez az új starthoz kapcsolódó három izoforma az új starthelyből, az 5. exonból és a változó hosszúságú 3′ UTR-ből áll. Érdekes módon, míg a GTEx és Sandor et al. (2017) nyilvánosan letöltött rövid olvasási adatai és a CAGE-csúcsadatok (FANTOM5) nem támasztották alá ezt az új starthelyet, a közelmúltbeli nyilvános NA12878 közvetlen RNS-adatsor4 csak egy SNCA-transzkriptet tartalmazott, amely megerősítette ezt az alternatív starthelyet. Továbbá, az 5. exon és az új starthely közötti új csomópontot az Intropolis rövid olvasási csomóponti adatok is megerősítették. Érdekes módon ez az új 5′ starthely az előrejelzések szerint új peptideket vezet be, miközben megtartja az olvasási keretet az 5. exonban.
Három SNCA-transzkriptet is azonosítottunk új véghelyekkel (3. ábra). Két izoforma (PB.1016.383, PB.1016.384) egy meghosszabbított 3′ UTR-t használt a 4. exonban, míg a harmadik izoforma (PB.1016.381) egy új 3′ exont használt a 4. intronban. Az újszerű utolsó exon és az előző exon közötti újszerű csomópontokat a nyilvános rövid leolvasási csomóponti adatok (Intropolis) alátámasztják. Az új 3′ UTR-ek csonka ORF-előrejelzést eredményeznek.
A teljes hosszúságú olvasatok normalizált számát az izoformák gyakoriságának helyettesítőjeként használva az egyik kanonikus SNCA izoformát (PB.1016.131) találtuk a legnagyobb gyakoriságúnak, 50-60%-os gyakorisággal az összes vizsgálati mintában (4. kiegészítő táblázat). A 41 izoformát tovább csoportosítottuk splicing-mintázatuk szerint (2. táblázat). Azok az izoformák, amelyek mind a hat exonnal rendelkeznek, a gyakoriság 95-97%-át teszik ki. Korábbi tanulmányok a DLB minták frontális kéregében a normálishoz képest a 3. exon hiányzó izoformák (SNCA126) kifejeződésének jelentős növekedését mutatták ki (Beyer et al., 2008); a mi összesített izoformaszámunk azt mutatja, hogy a DLB minták közül háromnál a normális mintákhoz képest kissé emelkedett a számszint, valamint az SNCA112 (5. exon kihagyása) variánsok PD és DLB esetében a normális mintákhoz képest.

Táblázat 2. táblázat. Az SNCA izoformák gyakorisága minden egyes minta esetében, splicing-minták szerint összesítve.
A teljes hosszúságú cDNS lehetővé teszi az izoforma-szintű fázisinformációt
A variánsok megnevezéséhez az SNP-ket cDNS használatával hívtuk meg, a 12 mintából származó összes teljes hosszúságú olvasatot felhalmozva (lásd a “Módszerek” című szakaszt). Összesen négy SNP-t hívtunk, és mindegyiket korábban annotáltuk a dbSNP-ben (3. táblázat, 3. ábra). A négy SNP mindegyike nem-CDS régiókban található, egy a 3′ UTR-ben (6. exon), egy a 4. intronban és kettő az 5′ UTR-ben (1. exon). A 3′ UTR SNP-t (chr4: 90646886) csak a legalább ~1 kb hosszú 3′ UTR-rel rendelkező izoformák fedik le, és ezért nem minden kanonikus izoforma fedezi ezt az SNP-t. Az intron 4 SNP-t (chr4: 90743331) csak az új, alternatív 3′ végű izoformák (PB.1016.383, PB.1016.384) fedik le, és nincs kapcsolatban a többi SNP-vel. A két 5′ UTR SNP-t (chr4: 90757312 és chr4: 90758389) két, egymást kölcsönösen kizáró exon 1 felhasználás fedi le, és ezért szintén nem kapcsolódnak egymáshoz.

3. táblázat. cDNS SNP információk.
A jelenlegi megközelítésünk csak a megfelelő lefedettségű átírt régiókban lévő szubsztitúciós variánsok hívására korlátozódik. A mi SNP-ink listájának összehasonlítása a hg19 dbSNP annotációval azt mutatja, hogy a kihagyott SNP-k vagy variánsok többsége vagy 1%-nál kisebb gyakoriságú volt a populációban, vagy nem egynukleotid szubsztitúció volt, vagy alacsony komplexitású régiókkal szomszédos. Például az rs77964369 (chr4: 90646532) a jelentések szerint 50/50 gyakoriságú T/A; ez a T azonban egy 11 genomiális As downstream szakasz szomszédságában van. Az Iso-Seq olvasathalmaz kézi vizsgálata, amely ~1 300 olvasatot tartalmaz ezen a helyen, nem utal a variációra, legalábbis a 12 mintánk között.
A mintaspecifikus olvasatok felhasználásával az egyes minták genotípusát minden egyes SNP-helyen megadjuk (3. táblázat). Azon kívül, hogy a PD-2 esetében túl kevés leolvasás van, és mind a négy SNP esetében nem meggyőző, a legtöbb más minta genotípusát meg tudtuk hívni. Figyelemre méltó, hogy a DLB-3 volt az egyetlen olyan minta, amely minden SNP-helyen heterozigóta. Egyébként nem figyeltünk meg semmilyen állapot-specifikus mintázatot az egyik genotípus előnyben részesítésére a másikkal szemben.
Diszkusszió
Az SNCA gén célzott dúsítását multiplex gDNS- és cDNS-könyvtárakon alkalmazó első tanulmányt írjuk le neurológiai betegségek tanulmányozására hosszú olvasású szekvenálással. A PacBio Sequel rendszer hosszú olvasási hossza megkönnyítette az SNCA gén teljes hosszúságú transzkript izoformák repertoárjának szekvenálását. Feltártuk az alternatív 5′ starthelyek és a változó 3′ UTR-hosszúságok használatának diverzitását, és megfigyeltük az ismert exon-kihagyási eseményeket, például az exon 3 deléciót (SNCA126) és az exon 5 deléciót (SNCA112). Ezenkívül a nagy 4-es intrononon belül új alternatív kezdő- és végpontokat azonosítottunk, amelyekről azt jósolták, hogy új fehérjékké transzlálódnak. Valószínű, hogy a célzott befogás nagy mélységű szekvenálási lefedettsége, a teljes transzkriptumok szekvenálásának képességével együtt lehetővé tette számunkra ezeknek a korábban le nem írt izoformáknak a kimutatását.
A különböző SNCA fehérjeizoformák biológiai és patológiai jelentőségét még nem sikerült teljes mértékben feltárni. A specifikus SNCA poszt-transzlációs módosítás és splicing izoformákat azonban összefüggésbe hozták az intracelluláris aggregációs hajlammal (Kalivendi és mtsai., 2010), és a humán szinukleinopátiákban eltérően expresszálódnak (Beyer és mtsai., 2008; Beyer és Ariza, 2012). Az SNCA poszt-transzlációs módosulásának vizsgálata azt mutatta, hogy a Lewy-testek, a szinukleinopátiák patológiai jellemzője, bőségesen tartalmaznak foszforilált, nitrált és monoubikvitinált SNCA-t (Kim és mtsai., 2014). A poszttranszkripcionális módosítás SNCA aggregációra gyakorolt hatását is vizsgálták. Az alternatív splicingről feltételezték, hogy befolyásolja az SNCA aggregációt. A 3. vagy az 5. exon deléciója funkcionális következményeket vetít előre: míg a 3. exon deléciója (SNCA126) az N-terminális fehérje-membrán interakciós domén megszakadásához vezet, ami kisebb aggregációt eredményezhet, addig az 5. exon deléciója (SNCA112) a strukturálatlan C-terminus jelentős lerövidülése miatt fokozott aggregációt eredményezhet (Lee és mtsai., 2001; Beyer, 2006). A DLB betegek frontális kéregében az SNCA112 szintje a kontrollokhoz képest jelentősen megnövekedett (Beyer és mtsai., 2008), míg a DLB betegek prefrontális kéregében az SNCA126 szintje csökkent (Beyer és mtsai., 2006). Ezzel szemben az SNCA126 expressziója emelkedettnek bizonyult a PD agyak frontális kéregében, és nem mutatott szignifikáns különbséget az MSA-ban (Beyer és mtsai., 2008). Az SNCA98 egy agyspecifikus splice-változat, amelyből hiányzik a 3. és az 5. exon, és eltérő expressziós szintet mutat a magzati és felnőtt agy különböző területein. Az SNCA98 overexpressziójáról számoltak be DLB, PD (Beyer et al., 2007) és MSA (Beyer et al., 2008) frontális kérgekben a kontrollokhoz képest. Ezenkívül az alternatív 3′UTR-használatot eredményező poszt-transzkripciós folyamatról azt jelentették, hogy hatással van az mRNS stabilitására és lokalizációjára (Fabian et al., 2010; Rhinn et al., 2012; Yeh és Yong, 2016). A különböző ismert SNCA fehérje izoformák aggregációs hajlamaira és a Lewy-testek összetételére vonatkozó további vizsgálatok indokoltak. Továbbá vizsgálatunk megalapozta a korábban ismert és új transzkriptek mRNS-kvantitatív elemzését egy nagyobb mintanagyságú, különböző klinikopatológiai stádiumú alanyokból álló mintában, az egyes alanyok több agyi régiójának felhasználásával. Az SNCA agyi régió-specifikus transzkriptomikai tájképének ezek az elemzései a neuropatológiai súlyosság összefüggésében informatívak lesznek a specifikus SNCA transzkript izoformák szerepét illetően a neuropatológiai stádiumok progressziójában és a Lewy-testek és Lewy-neuritok sűrűségének súlyosságában.
Ebben a tanulmányban egy szekvenálási és elemzési standard létrehozására összpontosítottunk az azonos alanyokból generált célzott gDNS és cDNS adatok elemzésére. Ez egy hatékony megközelítés, amely potenciálisan lehetővé teszi a gDNS-szekvenciák fázisait egy adott gén teljes régiójában a teljes hosszúságú transzkript izoformák szekvenciájának heterozigozitása alapján. A PacBio célzott gDNS-adatai ebben a vizsgálatban olyan fázist tartalmazó blokkokat eredményeztek, amelyek az SNCA központú 114 kb-os régió 81%-át lefedték, a leghosszabb fázist tartalmazó blokk pedig meghaladta az 54 kb-ot. Mivel a gDNS-fázist a leolvasás hossza és a heterozigozitás korlátozza, a növekvő leolvasáshossz valószínűleg nagyobb fázisblokkokat fog generálni.
A gDNS-variánselemzés megerősítette az ismert és azonosított új rövid tandemismétlődéseket (STR) az intronikus régiókban. Korábban például klónozással és Sanger-szekvenálással végzett fázisszekvenálással négy különböző haplotípust fedeztünk fel egy intronikus CT-gazdag régióban, amely változó ismétlődő szekvenciák egy klaszteréből állt (Lutz és mtsai., 2015). Kimutattuk, hogy egy specifikus haplotípus, amelyet 3. haplotípusnak neveztünk el, az Alzheimer-kóros betegeknél a Lewy-test patológiájának kialakulására vonatkozó kockázatot jelent. Itt validáltuk ennek az erősen polimorf, alacsony komplexitású régiónak a szekvenciáját és négy meghatározott haplotípusát. Bár a mintánk mérete kicsi volt, a “3. haplotípus” kizárólag a betegségben szenvedő betegeknél volt jelen (egy PD-beteg, két DLB-beteg), ami összhangban áll korábbi eredményeinkkel. A kísérleti eredmények és a korábbi publikációnk előfeltételezik, hogy a pontosan, azaz hosszú leolvasással definiált STR-ekkel és strukturális haplotípusokkal rendelkező synucleinopathiák asszociációs elemzéseit nagyobb mintaszámmal megismételjük.
Dolgozatunk bemutatta a PacBio Sequel rendszer képességét új teljes hosszúságú transzkriptek felfedezésére és egy betegségben szerepet játszó gén teljes teljes hosszúságú transzkript-repertoárjának jellemzésére. Továbbá azt is megmutattuk, hogy a hosszú olvasású gDNS pontosabban határozza meg a rövid szerkezeti variánsokat és a haplotípusokat, beleértve az STR-eket is, és ezáltal megkönnyítheti az SNP-ken kívüli, betegséggel összefüggő variánsok felfedezését és validálását. Összességében ez az új tudás rendkívül értékes és alkalmazható a komplex emberi betegségek, köztük az életkorral összefüggő neurodegeneratív rendellenességek, például a synucleinopathiák alapjául szolgáló genetikai etiológiák megértésének előmozdításában, amelyek a transzkript-tájkép perturbációjával járhatnak együtt.
Adatok elérhetősége
A gDNS három SMRT sejtjének nyers adatai elérhetőek a Zenodo.org oldalon, doi: 10.5281/zenodo.1560688. A cDNS nyers adatok egy SMRT cellája a Zenodo.org oldalon érhető el, doi: 10.5281/zenodo.1581809. A feldolgozott gDNS- és cDNS-eredmények, beleértve a gDNS-variánsokat és a cDNS-izoformákat is, a Zenodo.org címen érhetők el, doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F hozzájárult a vizsgálat koncepciójához és tervezéséhez. ET és WR megszervezte a szekvencia-adatbázisokat, elvégezte a szekvenciaelemzéseket, valamint elkészítette az összes ábrát és táblázatot. O-CG és JB kezelte az agyszöveteket és a nukleinminták előkészítését. TH generálta a szekvenálási adatsorokat. SK tervezte meg és szerezte be a reagenseket. OC-F, ET és WR írták a kézirat első tervezetét. OC-F szerezte a finanszírozást. Minden szerző hozzájárult a kézirat előkészítéséhez, elolvasta és jóváhagyta a benyújtott változatot.
Finanszírozás
Ezt a munkát részben a National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) finanszírozta.
Conflict of Interest Statement
ET, WR, TH és SK a Pacific Biosciences alkalmazottai vagy voltak a vizsgálat idején.
A többi szerző kijelenti, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségként értelmezhetők.
Megköszönések
Ez a kézirat előzetes nyomtatásként jelent meg a BioRxiv-on (Tseng et al, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Kiegészítő anyag
A cikkhez tartozó kiegészítő anyag online elérhető a következő címen: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Az öregedés és a neurodegeneratív rendellenességek arizonai tanulmányozása, valamint az agy- és testadományozási program. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-szinuklein szerkezet, poszttranszlációs módosítás és alternatív splicing mint aggregációfokozók. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Az alfa-szinuklein poszttranszlációs módosítása és az alternatív splicing mint a neurodegeneráció kiváltója. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., and Ariza, A. (2008). Az alfa-szinuklein, parkin és synphilin-1 izoformák differenciális expressziója Lewy-test betegségben. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Egy új alfa-szinuklein izoforma azonosítása és jellemzése, valamint szerepe a Lewy-testes betegségekben. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Alacsony alfa-szinuklein 126 mRNS-szint a Lewy-testes demenciában és az Alzheimer-kórban. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., and Filipowicz, W. (2010). Az mRNS transzláció és stabilitás szabályozása mikroRNS-ek által. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: többszörös szekvencia-illesztés nagy pontossággal és nagy áteresztőképességgel. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Oxidánsok indukálják az Α-szinuklein alternatív splicingjét: következmények a Parkinson-kórra nézve. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., and Halliday, G. M. (2014). Az alfa-szinuklein biológiája a Lewy-testes betegségekben. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Az AR-V9 androgénreceptor-variáns az AR-V7-gyel együtt expresszálódik prosztatarák metasztázisokban és előrejelzi az abirateron-rezisztenciát. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). A membránhoz kötött Α-szinuklein nagy aggregációs hajlandósággal rendelkezik, és képes a citoszolikus forma aggregációjának beindítására. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., and Chiba-Falek, O. (2015). Egy citozin-timin (CT)-gazdag haplotípus az SNCA 4-es intronjában a Lewy-test patológiájának kockázatát adja az Alzheimer-kórban és befolyásolja az SNCA expresszióját. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). A Lewy-testes demencia diagnózisa és kezelése: a DLB-konzorcium harmadik jelentése. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Jelentés a második Lewy-testes demencia nemzetközi workshopról: diagnózis és kezelés. A Lewy-testes demenciával foglalkozó konzorcium. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., and Isacson, O. (2012). A teljes hosszúságú alfa-szinuklein és három alternatív módon splicelt változatának transzkript expressziós szintjei a Parkinson-kór agyi régióiban és az alfa-szinuklein túlterjedésének transzgenikus egérmodelljében. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tízezer samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternatív Α-szinuklein transzkripthasználat mint konvergens mechanizmus a Parkinson-kór patológiájában. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). A tisztított, betegektől származó dopamin neuronok transzkriptomikai profilozása konvergens perturbációkat és a Parkinson-kór terápiáit azonosítja. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). A neurexin alternatív splicing kartográfiája egymolekulás hosszú leolvasású mRNS-szekvenálással feltérképezve. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). Az FMR1 splicing variánsok tájának megváltozott expressziója premutáció hordozókban. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). Az SNCA gén és a Parkinsonizmus közötti kapcsolat. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Az mRNS-ek alternatív poliadenilációja: A 3′-transzlálatlan régió számít a génexpresszióban. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar