- Introduzione
- Materiali e metodi
- Campioni di studio
- Estrazioni di DNA genomico e RNA
- Preparazione della biblioteca e sequenziamento
- Cattura del gDNA utilizzando IDT Xgen® Lockdown® Probes e sequenziamento a singola molecola
- Cattura del DNA utilizzando IDT Xgen® Lockdown® Probes e Isoform-Sequencing a singola molecola (Iso-Seq)
- Analisi gDNA
- Analisi delle varianti brevi e Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Analisi delle isoforme
- Isoform SNP Calling
- Risultati
- La cattura mirata del gDNA ha identificato variazioni note e nuove
- Targeted cDNA Capture identificato nuovi siti di inizio e fine
- Il cDNA a lunghezza intera consente informazioni di fasatura a livello di isoforma
- Discussione
- Data Availability
- Contributi degli autori
- Finanziamento
- Conflict of Interest Statement
- Riconoscimenti
- Materiale supplementare
Introduzione
I programmi trascrizionali e post-trascrizionali controllano i livelli di espressione genica e/o la produzione di più isoforme di mRNA distinte, e i cambiamenti in questi meccanismi portano alla disregolazione dell’espressione genica e a profili di espressione differenziali. Una regolazione trascrizionale e post-trascrizionale aberrante dei geni è abbondante nei tessuti del sistema nervoso umano e contribuisce alle differenze fenotipiche all’interno e tra gli individui in salute e in malattia.
La disregolazione dell’espressione dell’alfa-sinucleina è stata implicata nella patogenesi delle sinucleinopatie, in particolare nella malattia di Parkinson (PD) e nella demenza a corpi di Lewy e (DLB). Mentre il ruolo della sovraespressione SNCA nelle sinucleinopatie, principalmente PD, è stato ben stabilito, qui ci siamo concentrati sulla determinazione del repertorio completo delle isoforme di trascrizione SNCA in diverse sinucleinopatie. In precedenza, diverse diverse isoforme SNCA trascrizione sono stati descritti per SNCA gene, nato da splicing alternativo, siti di inizio trascrizione (TSSs), e la selezione dei siti di poliadenilazione (McLean et al., 2012; Xu et al., 2014). Lo splicing alternativo degli esoni codificanti dà origine a SNCA 140, SNCA 112, SNCA 126 e SNCA 98, dando origine a quattro isoforme proteiche (Beyer e Ariza, 2012). TSS alternativi del gene SNCA danno luogo a quattro diversi 5′UTR, e la selezione alternativa di diversi siti di poliadenilazione determina tre lunghezze principali del 3′UTR, senza impatto sulla composizione del prodotto proteico (Beyer e Ariza, 2012). Il nostro obiettivo generale è quello di acquisire nuove conoscenze sul contributo delle diverse specie di mRNA SNCA, noti e nuovi, alla patogenesi e l’eterogeneità di synucleinopathies.
Fino ad oggi, la maggior parte degli studi hanno utilizzato tecnologie di sequenziamento di lettura breve per interrogare la complessità trascrittoma nel cervello umano. La disponibilità di tecnologie di lettura lunga di terza generazione fornisce un quadro senza precedenti e quasi completo delle strutture delle isoforme. Tuttavia, il sequenziamento di trascrizione a lunga lettura esistente per i geni della malattia umana ha utilizzato un approccio basato su ampliconi (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Mentre questo approccio ha avuto successo nell’identificare lo splicing alternativo complesso nei geni della malattia umana, è limitato al disegno del primer PCR e non scoprirà siti alternativi di inizio e fine. Arricchimento mirato, come ad esempio attraverso l’uso di sonde IDT, può fornire vista completa isoforma di geni di interesse a basso costo di sequenziamento. Inoltre, altamente accurata lunghezza completa trascrizione legge consentire isoforma-specifica haplotyping.
Qui, presentiamo il primo studio conosciuto utilizzando la cattura mirata di gDNA e cDNA della regione del gene SNCA utilizzando PacBio SMRT sequenziamento. La regione del gene SNCA è lunga ~ 114 kb, composta da sei esoni con lunghezze di trascrizione intorno ai 3 kb. Abbiamo multiplexato 12 campioni di cervello umano da PD, DLB, e campioni di controllo normale e sequenziato la libreria di gDNA e cDNA sul sistema PacBio Sequel. Descriviamo le analisi bioinformatiche utilizzate per identificare SNPs, indel, e brevi ripetizioni tandem per la cattura gDNA, e haplotyping a livello di isoforma per i dati cDNA. Dimostriamo che la cattura mirata è un modo conveniente di studiare congiuntamente la variazione genomica e lo splicing alternativo in un gene neurale correlato alla malattia.
Materiali e metodi
Campioni di studio
La coorte di studio (N = 12) era costituita da individui con tre diagnosi neuropatologiche confermate dall’autopsia: (1) PD (N = 4); (2) DLB (N = 4); e (3) soggetti clinicamente e neuropatologicamente normali (N = 4). I tessuti cerebrali della corteccia frontale sono stati ottenuti attraverso la Kathleen Price Bryan Brain Bank (KPBBB) alla Duke University, il Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015), e Layton Aging and Alzheimer’s Disease Center alla Oregon Health and Science University. I fenotipi neuropatologici sono stati determinati nell’esame post mortem seguendo metodi standard consolidati seguendo il metodo e le raccomandazioni di pratica clinica di McKeith e colleghi (McKeith et al., 1999, 2005). La densità della patologia LB (in un insieme standard di regioni del cervello) ha ricevuto punteggi di lieve, moderata, grave e molto grave. I campioni di studio all’interno di ogni gruppo di diagnosi, PD e DLB, sono stati accuratamente selezionati in modo che la gravità dei fenotipi clinicopatologici fosse simile all’interno di ogni patologia. Tutti i cervelli hanno mostrato corpi di Lewy (LB) nel tronco encefalico, limbico e neocorticale, mentre il PD ha mostrato punteggi McKeith da gravi a molto gravi nella sub-nigra e nell’amigdala. Tutti i cervelli non indicano AD secondo i criteri CERAD e lo stadio di Braak e Braak = II. I campioni cerebrali neurologicamente sani sono stati ottenuti da tessuti post mortem di soggetti clinicamente normali che sono stati esaminati, nella maggior parte dei casi, entro 1 anno dalla morte e sono stati trovati per non avere disturbi cognitivi o parkinsonismo e risultati neuropatologici insufficienti per la diagnosi di PD, malattia di Alzheimer (AD), o altri disturbi neurodegenerativi. Tutti i campioni erano bianchi. Demografia e neuropatologia per questi soggetti sono riassunti nella Tabella 1 supplementare. Il progetto è stato approvato dal Duke Institution Review Board (IRB) che ha fornito un’approvazione etica. I metodi sono stati eseguiti in conformità con le linee guida e regolamenti pertinenti.
Estrazioni di DNA genomico e RNA
Il DNA genomico è stato estratto dai tessuti cerebrali con il protocollo standard Qiagen (Qiagen, Valencia, CA). RNA totale è stato estratto da campioni di cervello (100 mg) utilizzando TRIzol reagente (Invitrogen, Carlsbad, CA) seguita da purificazione con un kit RNeasy (Qiagen, Valencia, CA), seguendo il protocollo del produttore. gDNA e RNA concentrazione è stata determinata spettrofotometricamente, e la qualità dei campioni di RNA e la mancanza di degradazione significativa sono stati confermati da misure del numero di integrità RNA (RIN, Tabella supplementare 1) utilizzando un Bioanalyzer Agilent.
Preparazione della biblioteca e sequenziamento
Cattura del gDNA utilizzando IDT Xgen® Lockdown® Probes e sequenziamento a singola molecola
Circa 2 μg di ogni campione di gDNA è stato tagliato a 6 kb utilizzando il Covaris g-TUBE e legato con adattatori con codice a barre. Un pool equimolare di 12-plex libreria gDNA codificata a barre (2 μg totale) è stato inserito nella cattura basata su sonda con un pannello di geni SNCA progettato su misura.
Una libreria SMRTBell è stato costruito utilizzando 626 ng di gDNA catturato e ri-amplificato1.
Cattura del DNA utilizzando IDT Xgen® Lockdown® Probes e Isoform-Sequencing a singola molecola (Iso-Seq)
Circa 100-150 ng di RNA totale per reazione è stata trascritta inversa utilizzando il kit di sintesi di cDNA Clontech SMARTer e 12 oligo dT con codice a barre specifico del campione (con sequenze di codici a barre PacBio 16mer, vedi metodi supplementari). Tre reazioni di trascrizione inversa (RT) sono state elaborate in parallelo per ogni campione. L’ottimizzazione della PCR è stata utilizzata per determinare il numero di ciclo di amplificazione ottimale per le reazioni di PCR su larga scala a valle. Un singolo primer (primer IIA dal kit Clontech SMARTer 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) è stato utilizzato per tutte le reazioni PCR post-RT. I prodotti di PCR su larga scala sono stati purificati separatamente con 1X AMPure perline PB, e il bioanalizzatore è stato utilizzato per il QC. Un pool equimolare di 12-plex biblioteca cDNA con codice a barre (1 μg totale) è stato immesso nella cattura basata su sonda con un pannello personalizzato gene SNCA progettato.
Una libreria SMRTBell è stato costruito utilizzando 874 ng di catturato e ri-amplificato cDNA2. Una cella SMRT 1M (filmato di 6 ore) è stata sequenziata sulla piattaforma PacBio Sequel utilizzando la chimica 2.0.
Analisi gDNA
Il sequenziamento dei dati gDNA con codice a barre è stato eseguito su tre cellule SMRT 1M utilizzando la chimica 2.0. I dati sono stati demultiplexati eseguendo l’applicazione Demultiplex Barcodes in PacBio SMRT Link v6.0.
Analisi delle varianti brevi e Phasing
Le letture della Sequenza di Consenso Circolare (CCS) sono state generate utilizzando SMRT Analysis 6.0 da ogni set di dati demultiplexati e allineate al genoma di riferimento hg19 utilizzando minimap2. I duplicati PCR dall’amplificazione post-cattura sono stati identificati mediante la mappatura degli endpoint e contrassegnati utilizzando uno script personalizzato. Le varianti brevi sono state chiamate utilizzando GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Dopo un primo passaggio di filtraggio utilizzando la profondità di copertura e le metriche di qualità, le varianti sono state ispezionate manualmente in IGV3. Se le varianti non erano in fase con SNPs vicini, sono state filtrate manualmente. I siti delle varianti che hanno superato la cura manuale sono stati utilizzati insieme agli allineamenti CCS deduplicati per il read-backed phasing con WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Subsequenze allineate a chr4: 90742331-90742559 (hg19) sono stati estratti per ogni campione. Dopo aver ispezionato la distribuzione delle dimensioni di queste sottosequenze, esse sono state raggruppate per dimensione e similarità di sequenza usando una combinazione di python e MUSCLE (Edgar, 2004), e una sequenza di consenso è stata generata indipendentemente per ogni cluster.
Script personalizzati e flussi di lavoro ulteriormente descritti in https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Analisi delle isoforme
Il sequenziamento dei dati del cDNA con codice a barre è stato effettuato su una cella SMRT 1M sul sistema PacBio Sequel utilizzando la chimica 2.0. L’analisi bioinformatica è stata effettuata utilizzando l’applicazione IsoSeq3 in PacBio SMRT Analysis v6.0.0 per ottenere sequenze di alta qualità, isoforme di lunghezza completa (vedi Metodi supplementari per ulteriori informazioni).
Isoform SNP Calling
Le letture di lunghezza completa associate alle 41 isoforme finali da tutti i 12 campioni sono stati allineati al genoma hg19 per creare un pile-up. Le basi con QV inferiore a 13 sono state escluse. Poi, ad ogni posizione con almeno 40 basi di copertura, è stato applicato un test esatto di Fisher con correzione di Bonferroni con un cutoff p di 0,01. Solo gli SNP di sostituzione non vicini a regioni omopolimeriche (tratti di 4 o più dello stesso nucleotide) sono stati chiamati. Dopo la chiamata degli SNP, il genotipo per ogni campione è stato determinato contando il numero di letture full length (FL) specifiche del campione. Se un campione aveva 5+ letture FL che supportavano sia la base di riferimento che quella alternativa, era eterozigote. Se un campione aveva 5+ letture FL che supportavano un allele e meno di 5 letture FL per l’altro, era omozigote. Altrimenti, era inconcludente. Gli script sono disponibili presso: https://github.com/Magdoll/cDNA_Cupcake.
Risultati
Abbiamo progettato sonde personalizzate per il gene SNCA ed eseguito la cattura mirata di gDNA e cDNA su una libreria multiplexed composto da 12 campioni di cervello umano da PD, DLB, e controlli normali (Figura 1, Tabella supplementare 1). Le librerie di gDNA e cDNA sono state sequenziate sulla piattaforma PacBio Sequel. L’analisi bioinformatica è stata fatta utilizzando il software PacBio seguita da analisi personalizzata.
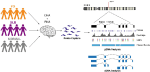
Figura 1. Presentazione schematica del disegno dello studio. Il DNA e l’RNA sono stati estratti da tessuti cerebrali postmortem di pazienti con malattia di Parkinson, demenza a corpi di Lewy e gruppi di controllo. Le librerie di gDNA e cDNA sono state realizzate utilizzando l’ibridazione di sonde e sequenziate sul sistema PacBio Sequel. L’analisi è stata eseguita utilizzando il software PacBio e altri strumenti esistenti.
La cattura mirata del gDNA ha identificato variazioni note e nuove
Dopo aver generato sequenze circolari di consenso (CCS) e rimosso i duplicati PCR (metodi supplementari), abbiamo ottenuto una copertura unica media da 16 a 71 volte della regione del gene SNCA. Le letture CCS avevano una lunghezza media dell’inserto di 2,9 kb e una precisione media di lettura del 98,9%. Con l’eccezione di una regione di 5 kb intenzionalmente scoperto da sonde a causa della presenza di elementi LINE (hg19 chr4: 90697216-90702113) e una regione 2,1 kb di alto contenuto di GC intorno esone 1, c’era una copertura sufficiente per genotipizzare entrambi gli aplotipi per ciascuno dei 212 campioni (Figura 2, Figura supplementare 1).

Figura 2. Cattura mirata del gDNA e fasatura. Un esempio che mostra un campione da ogni condizione. La traccia superiore mostra una delle isoforme SNCA, seguita dalla copertura gDNA per i tre campioni. La traccia delle varianti mostra ogni SNP e sono codificati a colori per eterozigoti (viola), omozigoti alternativi (arancione) e omozigoti di riferimento (grigio). I blocchi di fase sono mostrati in blu chiaro. Traccia inferiore mostra posizioni sonda di cattura. La regione di dropout nel disegno della sonda è dovuta a due elementi LINE nel mezzo dell’introne 4. Per la copertura gDNA e le informazioni di fasatura di tutti i 12 campioni, vedere le figure supplementari.
Usando GATK4 HC, filtraggio basato sulla qualità, e la cura manuale, abbiamo identificato 282 SNPs e 35 indel, tra cui 8 SNPS e 13 indel non trovato in dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (Tabella supplementare 2). Nessuna variante è stata identificata nella regione codificante per SNCA, anche se otto varianti sono state identificate nelle regioni non tradotte. La maggior parte delle varianti identificate, comprese diverse brevi ripetizioni tandem (STR), rientrano negli introni 2, 3 e 4.
Abbiamo precedentemente descritto una regione ricca di CT altamente polimorfica nell’introne 4 di SNCA con quattro aplotipi osservati (Lutz et al., 2015). Mentre questa regione altamente ripetitiva e strutturalmente variabile si è rivelata difficile da genotipizzare con GATK4 HC, siamo stati in grado di costruire sequenze di consenso per tutti i 12 campioni e osservato tutti e 4 gli aplotipi precedentemente scoperto (Figura supplementare 2). Inoltre, abbiamo identificato una nuova STR nell’introne 4 che consiste in un’unità di tre basi ripetuta 16 volte nel riferimento. All’interno dei 12 campioni, abbiamo identificato tre aplotipi, con 9, 12 e 15 copie dell’unità ripetuta TTG. GATK HC ha genotipizzato correttamente tutti questi tranne un aplotipo per PD-4, che aveva una copertura piuttosto bassa in questa regione. Tuttavia, con i dati forniti per questo campione, il genotipo può essere determinato tramite ispezione visiva (Tabella 1).

Tabella 1. Una ripetizione tandem tripletta romanzo nell’introne 4 (chr4: 90713442).
Abbiamo usato le varianti brevi rilevate da GATK HC in combinazione con lo strumento di fasatura basato sulla lettura WhatsHap (Martin et al., 2016) per fase le letture CCS attraverso il locus, con una gamma di successo guidato principalmente dalla densità di variante eterozigote sul locus. I campioni PD-1, PD-4, N-4, DLB-1, e DLB-4 avevano lunghi tratti di eterozigosi bassa, con pochi, brevi blocchi di fase, mentre gli altri campioni hanno prodotto blocchi di fase che vanno da 7 a 18 volte la lunghezza media della lettura, fino a 54 kb (Figura supplementare 3).
Targeted cDNA Capture identificato nuovi siti di inizio e fine
Abbiamo elaborato i dati PacBio cDNA (Iso-Seq) utilizzando il software PacBio SMRT Analysis. Dopo la mappatura dei dati Iso-Seq a hg19 e la rimozione degli artefatti (Tabella supplementare 3, Figura supplementare 4), abbiamo ottenuto un set finale di 41 isoforme SNCA (Figura 3). Tutte le isoforme finali hanno tutti i siti di splice canonici (GT-AG o GC-AG) e sono supportati da un totale di 20 o più letture a lunghezza intera. La maggior parte delle isoforme (28 su 41) hanno tutti e sei gli esoni, differendo solo nell’uso di siti di inizio alternativi 5′ e 3′ lunghezze UTR. Le lunghezze 3′ UTR variavano tra 300 e 2,6 kb. L’uso del sito di inizio 5′ alternativo altamente diversificato in SNCA è noto; ciò che è meno noto è la lunghezza variabile 3′ UTR, che era stato precedentemente studiato utilizzando RNA-seq dati che non hanno risolto le strutture full-length isoform (Rhinn et al., 2012). I dati Iso-Seq mostrano che la lunghezza variabile 3′ UTR sembra accoppiato con tutte le possibili combinazioni di siti di inizio 5′ senza accoppiamento preferenziale. Quasi nessuna della variabilità in inizio e fine sito cambia il telaio di lettura aperta previsto (Figura supplementare 5) e si prevede di tradurre alla sequenza canonica 141 aminoacidi.

Figura 3. Le isoforme SNCA catturate con Iso-Seq mirato identificano nuovi siti di inizio e fine. La maggior parte della complessità delle isoforme deriva dall’uso combinatorio di lunghezze alternative 3′ UTR e l’esone 1, con alcuni rari siti di giunzione alternativi trovati nell’esone 1 (verde), 2 (rosso) e 4 (blu). Tutte le giunzioni hanno siti di splice canonici. Abbiamo identificato cinque isoforme che hanno saltato l’esone 5 e due isoforme che hanno saltato l’esone 3. Abbiamo anche identificato nuovi siti di inizio (arancione) e fine (viola) nell’introne 4. Chiamato SNPs sono contrassegnati in viola.
Abbiamo ulteriormente convalidato il romanzo (ma canonico) giunzioni utilizzando pubblicamente disponibili dati di giunzione breve lettura. Il database Intropolis (v1, https://github.com/nellore/intropolis) combina oltre 21.000 RNA-seq pubblicamente disponibili. A causa dell’elevato volume di dati di giunzione supportati da una sola lettura breve, per questo studio, abbiamo bisogno di un minimo di 10 supporto di lettura breve (combinato da tutti i >21.000 set di dati RNA-seq) per confermare il nostro Iso-Seq giunzioni romanzo. Con l’eccezione delle nuove giunzioni per PB.1016.253 e PB.1016.296 (Figura 3), tutte le altre nuove giunzioni sono supportate dal set di dati Intropolis. È interessante notare che queste nuove giunzioni hanno un supporto significativamente inferiore rispetto alle giunzioni annotate da Gencode. Per esempio, le due giunzioni romanzo in PB.1016.139 introdotto dal romanzo esone sono supportati da 2.519 e 44 Intropolis conteggi di lettura breve, rispettivamente, mentre le altre quattro giunzioni noti sono supportati da oltre 1 milione di conteggi di lettura breve. Questo dimostra il potere dell’arricchimento mirato utilizzando il sequenziamento del trascrittoma completo per rilevare isoforme rare e nuove.
Abbiamo osservato due isoforme con salto dell’esone 3 (SNCA126) e cinque isoforme con salto dell’esone 5 (SNCA112). Ancora una volta, la diversità di splicing in questi due gruppi di salto dell’esone deriva principalmente dall’uso diverso di siti di inizio alternativi 5′ e dalla lunghezza variabile 3′ UTR. La predizione dell’ORF mostra che saltare l’esone 3 o l’esone 5 accorcia l’ORF ma mantiene la struttura di lettura. Tre isoforme hanno nuovi siti finali 3′ situati nell’introne 4.
Abbiamo identificato un sito di inizio 5′ precedentemente non annotato situato nell’introne 4 (hg19 chr4: 90692548-90693045, Figura 3). Le tre isoforme associate a questo nuovo inizio è costituito dal nuovo sito di inizio, esone 5, e variabile 3′ lunghezze UTR. È interessante notare che, mentre pubblicamente scaricato dati di lettura breve da GTEx e Sandor et al. (2017) e dati di picco CAGE (FANTOM5) non ha sostenuto questo sito di inizio romanzo, un recente pubblico NA12878 diretto RNA set di dati4 conteneva solo un trascritto SNCA che ha confermato questo sito di inizio alternativo. Inoltre, la nuova giunzione tra l’esone 5 e il nuovo sito di inizio è confermato da Intropolis breve leggere i dati di giunzione. È interessante notare che questo nuovo sito di inizio 5′ è previsto per introdurre nuovi peptidi pur mantenendo la struttura di lettura nell’esone 5.
Abbiamo anche identificato tre trascrizioni SNCA con nuovi siti finali (Figura 3). Due isoforme (PB.1016.383, PB.1016.384) hanno usato un 3′ UTR esteso nell’esone 4, mentre la terza isoforma (PB.1016.381) ha usato un nuovo esone 3′ nell’introne 4. Le nuove giunzioni tra l’ultimo esone romanzo e l’esone precedente sono supportate da dati pubblici di giunzione di lettura breve (Intropolis). Il romanzo 3′ UTR risultano in una predizione ORF troncata.
Usando il conteggio normalizzato full-length letto come proxy per l’abbondanza isoforma, troviamo una delle isoforme canoniche SNCA (PB.1016.131) per essere il più abbondante, con un’abbondanza del 50-60% in tutti i campioni soggetto (Tabella supplementare 4). Abbiamo ulteriormente raggruppato le 41 isoforme secondo i loro modelli di splicing (Tabella 2). Le isoforme che hanno tutti i sei esoni rappresentano il 95-97% dell’abbondanza. Studi precedenti hanno mostrato un marcato aumento dell’espressione delle isoforme mancanti dell’esone 3 (SNCA126) nella corteccia frontale dei campioni DLB rispetto al normale (Beyer et al., 2008); il nostro conteggio aggregato delle isoforme mostra che tre dei campioni DLB hanno un livello di conteggio leggermente elevato rispetto ai campioni normali così come le varianti SNCA112 (esone 5 skipping) per PD e DLB rispetto ai campioni normali.

Tabella 2. Abbondanza delle isoforme SNCA per ogni campione, aggregate in base ai modelli di splicing.
Il cDNA a lunghezza intera consente informazioni di fasatura a livello di isoforma
Abbiamo chiamato gli SNP utilizzando il cDNA accumulando tutte le letture a lunghezza intera dai 12 campioni per chiamare le varianti (vedi sezione “Metodi”). Un totale di quattro SNP sono stati chiamati e tutti erano precedentemente annotati in dbSNP (Tabella 3, Figura 3). I quattro SNPs sono tutti situati in regioni non-CDS, uno nel 3′ UTR (esone 6), uno nell’introne 4, e due nel 5′ UTR (esone 1). Il 3′ UTR SNP (chr4: 90646886) è coperto solo da isoforme con un 3′ UTR che è almeno ~ 1 kb lungo, e quindi, non tutte le isoforme canoniche coprire questo SNP. L’introne 4 SNP (chr4: 90743331) è coperto solo dalle nuove isoforme alternative 3′ end (PB.1016.383, PB.1016.384) e non è collegato a nessuno degli altri SNP. I due SNPs 5′ UTR (chr4: 90757312 e chr4: 90758389) sono coperti da due usi reciprocamente esclusivi dell’esone 1 e quindi non sono anche collegati.

Tabella 3. Informazioni SNP cDNA.
Il nostro approccio attuale è limitato a chiamare solo varianti di sostituzione nelle regioni trascritte con sufficiente copertura. Confrontando l’elenco dei nostri SNPs con l’annotazione hg19 dbSNP mostra che la maggior parte degli SNPs o delle varianti mancate erano o meno dell’1% di frequenza nella popolazione, non erano sostituzioni di singolo nucleotide, o adiacenti a regioni a bassa complessità. Per esempio, rs77964369 (chr4: 90646532) è segnalato per avere 50/50 di frequenza di T/A; tuttavia, questo T è adiacente a un tratto di 11 genomica As a valle. Ispezione manuale del pile-up di lettura Iso-Seq, che ha ~ 1.300 letture in questo sito, non suggerisce prove di variazione almeno tra i nostri 12 campioni.
Utilizzando le letture specifiche del campione, chiamiamo il genotipo di ogni campione in ogni posizione SNP (Tabella 3). Oltre a PD-2 che ha troppo poche letture ed è inconcludente per tutti e quattro gli SNP, siamo stati in grado di chiamare il genotipo per la maggior parte degli altri campioni. In particolare, DLB-3 è stato l’unico campione che è eterozigote in tutte le posizioni SNP. Altrimenti, non abbiamo osservato alcun modello condizione-specifico di preferire un genotipo all’altro.
Discussione
Descriviamo il primo studio utilizzando l’arricchimento mirato del gene SNCA su gDNA multiplexed e librerie cDNA per lo studio delle malattie neurologiche utilizzando sequenziamento lunga lettura. Le lunghezze di lettura del sistema PacBio Sequel hanno facilitato il sequenziamento del repertorio completo delle isoforme di trascrizione del gene SNCA. Abbiamo rivelato la diversità nell’uso di siti di inizio alternativi 5′ e lunghezze variabili 3′ UTR e osservato eventi noti esone skipping, come delezione esone 3 (SNCA126) e delezione esone 5 (SNCA112). Inoltre, sono stati identificati nuovi siti alternativi di inizio e fine all’interno del grande introne 4 che si prevede siano tradotti in nuove proteine. È probabile che l’alta profondità della copertura di sequenziamento della cattura mirata, in combinazione con la capacità di sequenziare trascrizioni complete, ci ha permesso di rilevare queste isoforme precedentemente non descritte.
Il significato biologico e patologico delle diverse isoforme della proteina SNCA deve ancora essere completamente scoperto. Tuttavia, specifiche isoforme SNCA posttraslazionali e di splicing sono state associate alla propensione all’aggregazione intracellulare (Kalivendi et al., 2010) e sono diversamente espresse nelle sinucleinopatie umane (Beyer et al., 2008; Beyer e Ariza, 2012). Gli studi sulla modifica post-trascrizionale di SNCA hanno mostrato che i corpi di Lewy, il segno distintivo patologico delle sinucleinopatie, contengono abbondantemente SNCA fosforilato, nitrato e monoubiquitinato (Kim et al., 2014). Sono stati studiati anche gli effetti della modifica post-trascrizionale sull’aggregazione SNCA. Lo splicing alternativo è stato suggerito per influenzare l’aggregazione SNCA. Una delezione dell’esone 3 o 5 predice conseguenze funzionali: mentre la delezione dell’esone 3 (SNCA126) porta all’interruzione del dominio N-terminale di interazione proteina-membrana che può portare ad una minore aggregazione, e la delezione dell’esone 5 (SNCA112) può portare ad una maggiore aggregazione a causa di un significativo accorciamento del C-terminale non strutturato (Lee et al., 2001; Beyer, 2006). Nella corteccia frontale della DLB, SNCA112 è aumentato notevolmente rispetto ai controlli (Beyer et al., 2008), mentre i livelli di SNCA126 sono diminuiti nella corteccia prefrontale dei pazienti DLB (Beyer et al., 2006). Al contrario, l’espressione di SNCA126 ha mostrato un aumento nella corteccia frontale dei cervelli di PD e nessuna differenza significativa nei MSA (Beyer et al., 2008). SNCA98 è una variante splice specifica per il cervello che manca di entrambi gli esoni 3 e 5 e mostra diversi livelli di espressione in varie aree del cervello fetale e adulto. La sovraespressione di SNCA98 è stata riportata nelle cortecce frontali di DLB, PD (Beyer et al., 2007), e MSA (Beyer et al., 2008) rispetto ai controlli. Inoltre, il processo post-trascrizionale con conseguente uso alternativo 3′UTR è stato segnalato per avere effetti sulla stabilità e localizzazione dell’mRNA (Fabian et al., 2010; Rhinn et al., 2012; Yeh e Yong, 2016). Ulteriori indagini riguardanti le propensioni all’aggregazione delle diverse isoforme note della proteina SNCA e la composizione dei corpi di Lewy sono giustificate. Inoltre, il nostro studio ha posto le basi per le analisi di quantificazione dell’mRNA dei trascritti precedentemente noti e nuovi in un campione più ampio composto da soggetti con una gamma di stadi clinicopatologici utilizzando diverse regioni del cervello di ogni soggetto. Queste analisi del paesaggio trascrittomico specifico della regione cerebrale di SNCA nel contesto della gravità neuropatologica saranno informative rispetto al ruolo delle isoforme specifiche del trascritto SNCA nella progressione degli stadi neuropatologici e nella gravità dei corpi di Lewy e della densità dei neuriti di Lewy.
In questo lavoro, ci siamo concentrati sulla creazione di uno standard di sequenziamento e analisi per analizzare dati mirati di gDNA e cDNA generati dagli stessi soggetti. Questo è un approccio potente che permette potenzialmente la fasatura delle sequenze gDNA attraverso la regione completa di un particolare gene basato su eterozigosi nella sequenza delle isoforme di trascrizione full-length. I dati PacBio gDNA mirati in questo studio prodotto blocchi fasato che copriva 81% della regione 114 kb centrata su SNCA, con il blocco più lungo fasato superiore a 54 kb. Come gDNA fasatura è limitata dalla lunghezza di lettura e eterozigosi, aumentando le lunghezze di lettura probabilmente generare più grandi blocchi di fase.
gDNA analisi delle varianti confermato noto e identificato romanzo brevi ripetizioni tandem (STRs) nelle regioni intronic. Per esempio, in precedenza, utilizzando il sequenziamento di fase per clonazione e sequenziamento Sanger, abbiamo scoperto quattro aplotipi distinti all’interno di una regione intronica ricca di CT che comprendeva un cluster di sequenze ripetitive variabili (Lutz et al., 2015). Abbiamo dimostrato che un aplotipo specifico, chiamato aplotipo 3, conferisce il rischio di sviluppare la patologia dei corpi di Lewy nei pazienti di Alzheimer. Qui, abbiamo convalidato la sequenza di questa regione altamente polimorfica a bassa complessità e i suoi quattro aplotipi definiti. Anche se il nostro campione era piccolo, l'”aplotipo 3″ era presente esclusivamente nei pazienti malati (un paziente PD, due pazienti DLB), in linea con i nostri risultati precedenti. I risultati pilota e la nostra precedente pubblicazione forniscono la premessa per ripetere le analisi di associazione delle sinucleinopatie con STR e aplotipi strutturali accuratamente definiti, cioè, da lunghe letture, utilizzando un campione di dimensioni maggiori.
Il nostro articolo ha dimostrato la capacità del sistema PacBio Sequel di scoprire nuovi trascritti a lunghezza intera e caratterizzare il repertorio completo dei trascritti a lunghezza intera di un gene coinvolto in una malattia. Inoltre, abbiamo anche dimostrato che lunghe letture gDNA definire più accuratamente brevi varianti strutturali e aplotipi tra cui STRs e da che può facilitare la scoperta e la convalida delle varianti associate alla malattia altro che SNPs. Collettivamente, questa nuova conoscenza è molto preziosa e applicabile per far progredire la nostra comprensione delle eziologie genetiche, che possono comportare perturbazioni nel paesaggio delle trascrizioni, alla base di malattie umane complesse, compresi i disturbi neurodegenerativi legati all’età come le sinucleinopatie.
Data Availability
Le tre cellule SMRT di dati grezzi gDNA sono disponibili su Zenodo.org con doi: 10.5281/zenodo.1560688. Una cella SMRT di dati grezzi di cDNA è disponibile su Zenodo.org con doi: 10.5281/zenodo.1581809. I risultati elaborati di gDNA e cDNA, comprese le varianti di gDNA e le isoforme di cDNA, sono disponibili su Zenodo.org con doi: 10.5281/zenodo.3261805.
Contributi degli autori
OC-F ha contribuito all’ideazione e alla progettazione dello studio. ET e WR hanno organizzato i database delle sequenze, eseguito le analisi di sequenziamento e preparato tutte le figure e le tabelle. O-CG e JB hanno gestito i tessuti cerebrali e le preparazioni dei campioni nucleici. TH ha generato i set di dati di sequenziamento. SK ha progettato e ottenuto i reagenti. OC-F, ET e WR hanno scritto la prima bozza del manoscritto. OC-F ha ottenuto finanziamenti. Tutti gli autori hanno contribuito alla preparazione del manoscritto, hanno letto e approvato la versione presentata.
Finanziamento
Questo lavoro è stato finanziato in parte dal National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS).
Conflict of Interest Statement
ET, WR, TH, e SK sono o erano dipendenti di Pacific Biosciences al momento dello studio.
I restanti autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
Riconoscimenti
Questo manoscritto è stato rilasciato come pre-print a BioRxiv (Tseng et al, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Materiale supplementare
Il materiale supplementare per questo articolo può essere trovato online a: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona studio di invecchiamento e disturbi neurodegenerativi e programma di donazione del cervello e del corpo. Neuropatologia 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Struttura di Α-sinucleina, modifica post-traslazionale e splicing alternativo come potenziatori di aggregazione. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., e Ariza, A. (2012). Alfa-sinucleina modifica post-traslazionale e splicing alternativo come un trigger per la neurodegenerazione. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., e Ariza, A. (2008). Espressione differenziale delle isoforme di alfa-sinucleina, parkina e synphilin-1 nella malattia dei corpi di Lewy. Neurogenetica 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., e Ariza, A. (2007). Identificazione e caratterizzazione di una nuova isoforma di alfa-sinucleina e il suo ruolo nelle malattie dei corpi di Lewy. Neurogenetica 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Bassi livelli di mRNA di alfa-sinucleina 126 nella demenza a corpi di Lewy e nella malattia di Alzheimer. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., e Filipowicz, W. (2010). Regolazione della traduzione mRNA e la stabilità da microRNA. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: allineamento di sequenze multiple con alta precisione e alta produttività. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., e Kalyanaraman, B. (2010). Ossidanti indurre splicing alternativo di Α-sinucleina: implicazioni per la malattia di Parkinson. Radicali liberi. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., e Halliday, G. M. (2014). Biologia dell’alfa-sinucleina nelle malattie dei corpi di Lewy. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). La variante del recettore degli androgeni AR-V9 è coespressa con AR-V7 nelle metastasi del cancro alla prostata e predice la resistenza all’abiraterone. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). La Α-sinucleina legata alla membrana ha un’alta propensione all’aggregazione e la capacità di seminare l’aggregazione della forma citosolica. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., e Chiba-Falek, O. (2015). Un aplotipo ricco di citosina-timina (CT) nell’introne 4 di SNCA conferisce il rischio per la patologia dei corpi di Lewy nella malattia di Alzheimer e influenza l’espressione SNCA. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: veloce e preciso read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosi e gestione della demenza a corpi di Lewy: terzo rapporto del consorzio DLB. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Relazione del secondo workshop internazionale sulla demenza a corpi di Lewy: diagnosi e trattamento. Consorzio sulla demenza a corpi di Lewy. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., and Isacson, O. (2012). Livelli di espressione della trascrizione dell’alfa-sinucleina completa e delle sue tre varianti con splicing alternativo nelle regioni del cervello della malattia di Parkinson e in un modello murino transgenico di sovraespressione dell’alfa-sinucleina. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling scoperta accurata variante genetica a decine di migliaia di campioni. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Uso alternativo della trascrizione di Α-sinucleina come meccanismo convergente nella patologia della malattia di Parkinson. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Profilo trascrittomico di neuroni dopaminergici purificati derivati da pazienti identifica perturbazioni convergenti e terapeutiche per la malattia di Parkinson. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartografia di splicing alternativo neurexina mappato da singola molecola long-read mRNA sequencing. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). Il paesaggio dei trascritti SNCA attraverso le sinucleinopatie: nuove intuizioni dall’analisi di sequenziamento di lunghe letture. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., e Tassone, F. (2017). Espressione alterata del paesaggio delle varianti di splicing FMR1 nei portatori di premutazione. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). Il legame tra il gene SNCA e il parkinsonismo. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., e Yong, J. (2016). Poliadenilazione alternativa di mRNA: 3′-untranslated region conta nell’espressione genica. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar