- Introduction
- Materiais e Métodos
- Amostras de Estudo
- Extratos de DNA genômico e RNA
- Preparação e Sequenciamento de Bibliotecas
- GDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
- cDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Isoform-Sequencing (Iso-Seq)
- Análise gDNA
- Análise de Variante Curta e Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Análise de isoforma
- Isoform SNP Calling
- Resultados
- Captura de gDNA Identificado Identificado e Variações de Novel Alvo
- Target cDNA Capture Identified Novel Start and End Sites
- CDNA de comprimento total Permite informações de fase de Isoform-Level Phasing Information
- Discussão
- Dados Disponibilidade
- Author Contributions
- Funding
- Conflict of Interest Statement
- Agradecimentos
- Material Suplementar
Introduction
Programas transcríticos e pós-transcríticos controlam níveis de expressão gênica e/ou produção de múltiplas isoformas distintas de mRNA, e mudanças nestes mecanismos resultam em desregulação da expressão gênica e perfis diferenciais de expressão. A regulação gênica transcripcional e pós-transcricional aberta é abundante nos tecidos do sistema nervoso humano e contribui para diferenças fenotípicas dentro e entre indivíduos em saúde e doença.
Disregulação da expressão da alfa-sinucleína tem sido implicada na patogênese das sinucleinopatias, em particular a Doença de Parkinson (DP) e Demência com corpos de Lewy e (DLB). Enquanto o papel da superexpressão da SNCA nas sinucleinopatias, principalmente a DP, tem sido bem estabelecido, aqui focamos na determinação do repertório completo das isoformas de transcrição da SNCA em diferentes sinucleinopatias. Anteriormente, várias isoformas diferentes de transcrição SNCA foram descritas para o gene SNCA, surgidas a partir de emendas alternativas, sítios de início transcripcional (SSTs) e seleção de sítios de poladenilação (McLean et al., 2012; Xu et al., 2014). A emenda alternativa dos exões de codificação dá origem a SNCA 140, SNCA 112, SNCA 126 e SNCA 98, resultando em quatro isoformas proteicas (Beyer e Ariza, 2012). TSSs alternativos do gene SNCA resultam em quatro diferentes 5′UTRs, e a seleção alternativa de diferentes sites de poliadenilação determina três grandes comprimentos do 3′UTR, sem impacto na composição do produto protéico (Beyer e Ariza, 2012). Nosso objetivo principal é obter novos conhecimentos sobre a contribuição das diferentes espécies de mRNA SNCA, conhecidas e inovadoras, para a patogênese e heterogeneidade das sinucleinopatias.
Até o momento, a maioria dos estudos tem usado tecnologias de sequenciamento de leitura curta para interrogar a complexidade do transcriptoma no cérebro humano. A disponibilidade de tecnologias de terceira geração de leitura longa fornece uma imagem sem precedentes e quase completa das estruturas isoforma. No entanto, o sequenciamento de transcrição de leitura longa existente para genes de doenças humanas tem usado uma abordagem baseada em amplicon (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Embora esta abordagem tenha sido bem sucedida na identificação de emendas alternativas complexas em genes de doenças humanas, está limitada ao desenho do primer de PCR e não irá descobrir locais alternativos de início e fim. O enriquecimento específico, tal como através do uso de sondas de IDT, pode proporcionar uma visão isoforma abrangente dos genes de interesse a baixo custo de sequenciação. Além disso, a transcrição altamente precisa de leitura completa permite a haplotipagem específica de isoformas.
Aqui, apresentamos o primeiro estudo conhecido usando a captura orientada de gDNA e cDNA da região do gene SNCA usando o seqüenciamento PacBio SMRT. A região do gene SNCA tem um comprimento de ~114 kb, consistindo de seis exons com comprimento de transcrição de cerca de 3 kb. Nós multiplexamos 12 amostras de cérebro humano de PD, DLB e amostras de controle normal e sequenciamos a biblioteca de gDNA e cDNA no sistema PacBio Sequel. Descrevemos as análises bioinformáticas usadas para identificar SNPs, indels e repetições curtas em tandem para a captura de gDNA, e haplotipagem em nível de isoforma para os dados de cDNA. Mostramos que a captura orientada é uma forma econômica de estudar conjuntamente a variação genômica e emendas alternativas em um gene neural relacionado à doença.
Materiais e Métodos
Amostras de Estudo
A coorte do estudo (N = 12) consistiu de indivíduos com três diagnósticos neuropatológicos confirmados por autópsia: (1) DP (N = 4); (2) DLB (N = 4); e (3) indivíduos clínica e neuropatologicamente normais (N = 4). Os tecidos do córtex frontal foram obtidos através do Kathleen Price Bryan Brain Bank (KPBBB) na Duke University, do Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) e do Layton Aging and Alzheimer’s Disease Center na Oregon Health and Science University. Os fenótipos neuropatológicos foram determinados no exame post-mortem seguindo métodos padrão bem estabelecidos, seguindo as recomendações de método e prática clínica de McKeith e colegas (McKeith et al., 1999, 2005). A densidade da patologia LB (em um conjunto padrão de regiões cerebrais) recebeu escores de leve, moderado, severo e muito severo. As amostras do estudo dentro de cada grupo de diagnóstico, DP e DLB, foram cuidadosamente selecionadas de modo que a gravidade dos fenótipos clinicopatológicos fosse semelhante dentro de cada patologia. Todos os cérebros exibiram tronco cerebral, corpo límbico e neocortical Lewy (LBs), enquanto a DP mostrou escores de McKeith graves a muito graves na sub-nigra e na amígdala. Todos os cérebros indicam ausência de AD de acordo com os critérios do CERAD e estágio Braak e Braak = II. As amostras de cérebro neurologicamente saudáveis foram obtidas de tecidos pós-morte de indivíduos clinicamente normais que foram examinados, na maioria dos casos, dentro de 1 ano após a morte e que não apresentaram distúrbio cognitivo ou parkinsonismo e achados neuropatológicos insuficientes para o diagnóstico de DP, doença de Alzheimer (DA), ou outras doenças neurodegenerativas. Todas as amostras eram brancas. Os dados demográficos e neuropatológicos para estes sujeitos estão resumidos na Tabela Complementar 1. O projeto foi aprovado pelo Duke Institution Review Board (IRB), que forneceu uma aprovação ética. Os métodos foram realizados de acordo com as diretrizes e regulamentos pertinentes.
Extratos de DNA genômico e RNA
O DNA genômico foi extraído dos tecidos cerebrais pelo protocolo padrão Qiagen (Qiagen, Valência, CA). O RNA total foi extraído de amostras cerebrais (100 mg) usando reagente TRIzol (Invitrogen, Carlsbad, CA) seguido de purificação com um kit RNeasy (Qiagen, Valencia, CA), seguindo o protocolo do fabricante. A concentração de gDNA e RNA foi determinada espectrofotometricamente, e a qualidade das amostras de RNA e a ausência de degradação significativa foram confirmadas por medições do Número de Integridade do RNA (RIN, Tabela Suplementar 1) usando um Bioanalisador Agilente.
Preparação e Sequenciamento de Bibliotecas
GDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
Aproximadamente 2 μg de cada amostra de gDNA foi tosquiada a 6 kb usando o Covaris g-TUBE e ligada com adaptadores de código de barras. Um pool equimolar de 12 gDNA codificados com código de barras (2 μg total) foi inserido na captura baseada em sonda com um painel genético SNCA projetado sob medida.
Uma biblioteca SMRTBell foi construída usando 626 ng de gDNA1 capturado e reamplificado.
cDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Isoform-Sequencing (Iso-Seq)
Sobre 100-150 ng de RNA total por reacção foi transcrito ao contrário utilizando o kit de síntese cDNA Clontech SMARTer e 12 amostras de oligo dT com código de barras específico (com sequências de código de barras PacBio 16mer, ver Métodos Suplementares). Três reações de transcrição reversa (RT) foram processadas em paralelo para cada amostra. A optimização PCR foi utilizada para determinar o número óptimo do ciclo de amplificação para as reacções PCR de larga escala a jusante. Um único iniciador (iniciador IIA do kit Clontech SMARTer 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) foi utilizado para todas as reacções de PCR pósRT. Os produtos de PCR em grande escala foram purificados separadamente com contas PB 1X AMPure, e o bioanalisador foi utilizado para CQ. Um pool equimolar da biblioteca de cDNA com código de barras 12-plex (1 μg total) foi introduzido na captura baseada em sondas com um painel genético SNCA projetado sob medida.
A biblioteca SMRTBell foi construída usando 874 ng de cDNA2 capturado e reamplificado. Uma célula SMRT 1M (filme de 6 h) foi sequenciada na plataforma PacBio Sequel utilizando a química 2.0.
Análise gDNA
Sequenciação dos dados gDNA com código de barras foi executada em três células SMRT 1M utilizando a química 2.0. Os dados foram desmultiplexados executando a aplicação Demultiplex Barcodes no PacBio SMRT Link v6.0.
Análise de Variante Curta e Phasing
Sequência de Consenso Circular (CCS) foram geradas utilizando a Análise 6.0 da SMRT a partir de cada conjunto de dados desmultiplexados e alinhados com o genoma de referência hg19 utilizando o minimap2. As duplicatas PCR da amplificação pós-captura foram identificadas através do mapeamento de pontos finais e marcadas usando um script personalizado. Variantes curtas foram chamadas usando o GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Após uma primeira passagem de filtragem usando profundidade de cobertura e métricas de qualidade, as variantes foram inspecionadas manualmente no IGV3. Se as variantes não foram faseadas com SNPs próximos, elas foram filtradas manualmente. As variantes que passaram na cura manual foram utilizadas em conjunto com os alinhamentos CCS deduplicados para a fase de read-backed com WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Subsequences aligned to chr4: 90742331-90742559 (hg19) foram extraídas para cada amostra. Após inspecionar a distribuição de tamanho destas subseqüências, elas foram agrupadas por tamanho e similaridade de seqüência usando uma combinação de pitão e MUSCLE (Edgar, 2004), e uma seqüência consensual foi gerada independentemente para cada cluster.
Criptografias e fluxos de trabalho personalizados descritos em https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Análise de isoforma
Sequenciamento dos dados de cDNA codificados com código de barras foi em uma célula SMRT 1M no sistema PacBio Sequel usando a química 2.0. A análise bioinformática foi feita utilizando a aplicação IsoSeq3 na Análise PacBio SMRT v6.0.0 para obter sequências de isoformas completas de alta qualidade (ver Métodos Suplementares para mais informações).
Isoform SNP Calling
Leituras de comprimento total associadas às 41 isoformas finais de todas as 12 amostras foram alinhadas ao genoma hg19 para criar uma pilha. Bases com QV inferior a 13 foram excluídas. Então, em cada posição com pelo menos 40 bases de cobertura, um teste exato de Fisher com correção de Bonferroni é aplicado com um corte de p de 0,01. Apenas os SNPs de substituição não próximos a regiões homopoliméricas (trechos de 4 ou mais do mesmo nucleotídeo) foram chamados. Após a chamada do SNP, o genótipo para cada amostra foi determinado através da contagem do número de leituras de comprimento total (FL) específicas da amostra de apoio. Se uma amostra tinha mais de 5 leituras de FL suportando tanto a base de referência como a base alternativa, ela era heterozigótica. Se uma amostra tinha 5+ leituras de FL que suportavam um alelo e menos de 5 leituras de FL para o outro, era homozigoto. Caso contrário, era inconclusivo. Os scripts estão disponíveis em: https://github.com/Magdoll/cDNA_Cupcake.
Resultados
Concebemos sondas personalizadas para o gene SNCA e realizámos a captura direccionada de gDNA e cDNA numa biblioteca multiplexada constituída por 12 amostras de cérebro humano de PD, DLB, e controlos normais (Figura 1, Tabela Suplementar 1). As bibliotecas de gDNA e cDNA foram sequenciadas na plataforma PacBio Sequel. A análise bioinformática foi feita usando o software PacBio seguido pela análise personalizada.
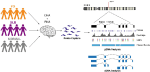
Figura 1. Apresentação esquemática do desenho do estudo. Os materiais de DNA e RNA foram extraídos de tecidos cerebrais pós-morte de pacientes com doença de Parkinson, Demência com Lewy Body e grupos de controle. As bibliotecas de gDNA e cDNA foram feitas usando hibridação de sonda e sequenciadas no sistema PacBio Sequel. A análise foi realizada utilizando o software PacBio e outras ferramentas existentes.
Captura de gDNA Identificado Identificado e Variações de Novel Alvo
Após a geração de seqüências de consenso circulares (CCS) e remoção de duplicatas de PCR (Métodos Suplementares), obtivemos uma cobertura média de 16 a 71 vezes a cobertura única da região do gene SNCA. As leituras de CCS tiveram um comprimento médio de inserção de 2,9 kb e uma precisão média de leitura de 98,9%. Com exceção de uma região de 5 kb intencionalmente descoberta por sondas devido à presença de elementos LINE (hg19 chr4: 90697216-90702113) e uma região de 2,1 kb de alto conteúdo de GC em torno do exon 1, houve cobertura suficiente para genotipar ambos os haplótipos para cada uma das 212 amostras (Figura 2, Figura Suplementar 1).

Figura 2. Captura e faseamento do gDNA direcionado. Um exemplo mostrando uma amostra de cada condição. A faixa superior mostra uma das isoformas SNCA, seguida da cobertura de gDNA para as três amostras. A faixa variante mostra cada SNP e são codificadas por cores para heterozigotos (roxo), homozigotos alternativos (laranja), e homozigotos de referência (cinza). Os blocos faseados são mostrados em azul claro. A faixa inferior mostra a localização das sondas de captura. A região de queda no desenho da sonda é devido a dois elementos de LINHA no meio do intron 4. Para a cobertura do gDNA e informação de faseamento de todas as 12 amostras, ver Figuras Suplementares.
Usando GATK4 HC, filtragem baseada na qualidade, e cura manual, identificamos 282 SNPs e 35 indels, incluindo 8 SNPS e 13 indels não encontrados no dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (Tabela Suplementar 2). Não foram identificadas variantes na região de codificação para SNCA, embora oito variantes tenham sido identificadas em regiões não traduzidas. A maioria das variantes identificadas, incluindo várias repetições curtas em tandem (STR), enquadram-se nos introns 2, 3 e 4,
Descrevemos anteriormente uma região altamente polimórfica rica em TC no intron 4 da SNCA com quatro haplótipos observados (Lutz et al., 2015). Embora esta região altamente repetitiva e estruturalmente variável tenha se mostrado difícil de genotipar com o HC GATK4, fomos capazes de construir seqüências de consenso para todas as 12 amostras e observamos todos os 4 haplótipos previamente descobertos (Figura Complementar 2). Além disso, identificamos um novo STR no intron 4 que consiste em uma unidade trifásica repetida 16 vezes na referência. Dentro das 12 amostras, identificamos três haplótipos, com 9, 12 e 15 cópias da unidade de repetição TTG. O GATK HC genotipou corretamente todos eles, exceto um haplótipo para PD-4, que teve cobertura bastante baixa nesta região. Entretanto, com os dados fornecidos para esta amostra, o genótipo pode ser determinado pela inspeção visual (Tabela 1).

Table 1. Uma nova repetição tandem em tandem no intron 4 (chr4: 90713442).
Usamos as variantes curtas detectadas pelo GATK HC em conjunto com a ferramenta de faseamento baseada em leitura WhatsHap (Martin et al., 2016) para fasear a leitura CCS em todo o locus, com uma gama de sucesso impulsionada principalmente pela densidade de variantes heterozigotas sobre o locus. As amostras PD-1, PD-4, N-4, DLB-1 e DLB-4 tinham longos trechos de baixa heterozigosidade, com muito poucos blocos de fase curtos, enquanto as outras amostras produziam blocos de fase que variavam de 7 a 18 vezes o comprimento médio lido, até 54 kb (Figura Suplementar 3).
Target cDNA Capture Identified Novel Start and End Sites
Processamos os dados de cDNA do PacBio (Iso-Seq) usando o software PacBio SMRT Analysis. Após mapear os dados Iso-Seq para hg19 e remover os artefatos (Tabela Complementar 3, Figura Complementar 4), obtivemos um conjunto final de 41 isoformas SNCA (Figura 3). Todas as isoformas finais têm todos os sites de emenda canônica (GT-AG ou GC-AG) e são suportadas por um total de 20 ou mais leituras completas. A maioria das isoformas (28 de 41) tem todos os seis exons, diferindo apenas no uso de sites de início alternativos 5′ e 3′ comprimentos UTR. Os comprimentos UTR 3′ variaram entre 300 e 2,6 kb. É conhecida a utilização de sites de início alternativos 5′ em SNCA; o que é menos conhecido é a variável 3′ comprimento UTR, que tinha sido previamente estudada utilizando dados RNA-seq que não resolviam estruturas de isoformas de comprimento total (Rhinn et al., 2012). Os dados da Iso-Seq mostram que a variável 3′ Comprimento UTR parece estar emparelhada com todas as combinações possíveis de sites de início 5′ sem acoplamento preferencial. Quase nenhuma das variações no local inicial e final altera a estrutura de leitura aberta prevista (Figura Complementar 5) e está prevista a tradução para a sequência canónica 141 aminoácidos.

Figura 3. As isoformas SNCA capturadas usando o Iso-Seq identifica novos locais de início e fim. A maioria da complexidade da isoforma vem do uso combinatório de comprimentos alternativos 3′ UTR e exon 1, com alguns raros sites alternativos de emenda encontrados no exon 1 (verde), 2 (vermelho), e 4 (azul). Todas as junções têm sítios de emenda canônica. Identificamos cinco isoformas que pularam o exon 5 e duas isoformas que pularam o exon 3. Também identificamos novos locais de início (laranja) e de fim (roxo) no intron 4. Os chamados SNPs são marcados em roxo.
Validamos ainda mais as junções novas (mas canônicas) usando dados de junção de leitura curta disponíveis publicamente. A base de dados Intropolis (v1, https://github.com/nellore/intropolis) combina mais de 21.000 RNA-seq. Devido ao alto volume de dados de junção suportados por apenas uma única leitura curta, para este estudo, nós exigimos um mínimo de 10 suportes de leitura curta (combinados de todos os conjuntos de dados >21.000 RNA-seq) para confirmar nossos novos junções Iso-Seq. Com exceção dos novos cruzamentos para PB.1016.253 e PB.1016.296 (Figura 3), todos os outros cruzamentos novos são suportados pelo conjunto de dados da Intropolis. Curiosamente, esses novos cruzamentos têm um suporte de leitura significativamente menos curto do que os cruzamentos anotados por Gencode-anotados. Por exemplo, os dois novos cruzamentos em PB.1016.139 introduzidos pelo novo exon são suportados por 2.519 e 44 contagens de leitura curta Intropolis, respectivamente, enquanto os outros quatro cruzamentos conhecidos são suportados por mais de 1 milhão de contagens de leitura curta. Isso mostra o poder do enriquecimento direcionado usando o sequenciamento de transcriptomas de comprimento total para detectar isoformas raras e novas.
Observamos duas isoformas com exon 3 skipping (SNCA126) e cinco isoformas com exon 5 skipping (SNCA112). Mais uma vez, a diversidade de emendas nestes dois grupos de saltos exon provém principalmente do uso diversificado de sites de início alternativos 5′ e variável 3′ UTR comprimento. A previsão ORF mostra que saltar exon 3 ou exon 5 encurta o ORF mas mantém o quadro de leitura. Três isoformas têm novos sites finais 3′ localizados no intron 4. A previsão ORF mostra que isto resulta em produto protéico truncado.
Identificamos um site de início previamente não anotado 5′ localizado no intron 4 (hg19 chr4: 90692548-90693045, Figura 3). As três isoformas associadas a este novo início consistem no novo site de início, exon 5, e na variável 3′ comprimentos UTR. Curiosamente, enquanto que os dados de leitura curta de GTEx e Sandor et al. (2017) e dados de pico CAGE (FANTOM5) não suportaram este novo site de início, um recente conjunto de dados públicos de RNA direto NA128784 continha apenas uma transcrição SNCA que confirmava este site de início alternativo. Além disso, a nova junção entre o exon 5 e o novo local de início é confirmada pelos dados de junção de leitura curta da Intropolis. Curiosamente, este novo site 5′ start site está previsto para introduzir novos peptídeos enquanto mantém o quadro de leitura no exon 5.
Nós também identificamos três transcrições SNCA com novos sites finais (Figura 3). Duas isoformas (PB.1016.383, PB.1016.384) usaram um UTR estendido 3′ no exon 4, enquanto a terceira isoforma (PB.1016.381) usou um novo 3′ exon no intron 4. As novas junções entre a última exon da novela e a exon anterior são suportadas por dados de junção de leitura curta pública (Intropolis). A novela 3′ UTRs resulta em uma previsão ORF truncada.
Usando a contagem normalizada de leitura de comprimento total como um proxy para abundância de isoformas, encontramos uma das isoformas SNCA canônicas (PB.1016.131) como a mais abundante, com uma abundância de 50-60% em todas as amostras sujeitas (Tabela Suplementar 4). Agrupamos ainda as 41 isoformas pelos seus padrões de emenda (Tabela 2). As isoformas que têm todos os seis exons são responsáveis por 95-97% da abundância. Estudos anteriores mostraram um aumento acentuado da expressão de isoformas faltando exon 3 (SNCA126) no córtex frontal de amostras DLB em comparação com amostras normais (Beyer et al., 2008); nossa contagem agregada de isoformas mostra que três das amostras DLB têm um nível de contagem ligeiramente elevado em comparação com as amostras normais, bem como as variantes SNCA112 (exon 5 skipping) para PD e DLB contra amostras normais.

Tabela 2. SNCA isoforma abundância para cada amostra, agregada por padrões de emenda.
CDNA de comprimento total Permite informações de fase de Isoform-Level Phasing Information
Chamamos SNPs usando cDNA empilhando todas as leituras de comprimento total das 12 amostras para chamar variantes (ver Seção “Métodos”). Um total de quatro SNPs foram chamados e todos foram previamente anotados no dbSNP (Tabela 3, Figura 3). Os quatro SNPs estão todos localizados em regiões não-CDS, um no 3′ UTR (exon 6), um no intron 4, e dois no 5′ UTR (exon 1). O 3′ UTR SNP (chr4: 90646886) só é coberto por isoformas com um 3′ UTR que tenha pelo menos ~1 kb de comprimento, pelo que nem todas as isoformas canónicas cobrem este SNP. O intron 4 SNP (chr4: 90743331) só é coberto pela nova alternativa 3′ end isoforms (PB.1016.383, PB.1016.384) e não está ligado a nenhum dos outros SNPs. Os dois 5′ UTR SNPs (chr4: 90757312 e chr4: 90758389) são cobertos por dois usos exon 1 mutuamente exclusivos e, portanto, também não estão ligados.

Table 3. cDNA SNP information.
A nossa abordagem atual limita-se a chamar apenas variantes de substituição em regiões transcritas com cobertura suficiente. Comparando a lista de nossos SNPs com a anotação hg19 dbSNP mostra que a maioria dos SNPs ou variantes ausentes eram menos de 1% de freqüência na população, não eram substituições de nucleotídeos isolados, ou adjacentes a regiões de baixa complexidade. Por exemplo, rs77964369 (chr4: 90646532) é relatado ter 50/50 de frequência de T/A; entretanto, este T é adjacente a um trecho de 11 genômicos As downstream. A inspeção manual da pilha de leitura Iso-Seq, que tem ~1.300 leituras neste local, não sugere evidência de variação pelo menos entre nossas 12 amostras.
Usando as leituras específicas da amostra, chamamos o genótipo de cada amostra em cada local do SNP (Tabela 3). Além do PD-2 ter muito poucas leituras e ser inconclusivo para todos os quatro SNPs, fomos capazes de chamar o genótipo para a maioria das outras amostras. Notavelmente, DLB-3 foi a única amostra que é heterozigota em todas as localizações SNP. Caso contrário, não observamos nenhum padrão específico de condição de preferir um genótipo a outro.
Discussão
Descrevemos o primeiro estudo usando enriquecimento direcionado do gene SNCA em bibliotecas de gDNA e cDNA multiplexados para o estudo de doenças neurológicas usando sequenciamento de leitura longo. Os longos comprimentos de leitura do sistema PacBio Sequel facilitaram o seqüenciamento do repertório de isoformas de transcrição completa do gene SNCA. Revelamos a diversidade no uso de sites de início alternativos 5′ e da variável 3′ comprimentos UTR e observamos eventos conhecidos de exon pulando, tais como exon 3 deleção (SNCA126) e exon 5 deleção (SNCA112). Além disso, foram identificados novos locais alternativos de início e fim dentro do grande intron 4 que estão previstos para serem traduzidos para novas proteínas. É provável que a alta profundidade da cobertura sequencial da captura dirigida, em combinação com a capacidade de sequenciar transcrições completas, nos permitiu detectar estas isoformas previamente não descritas.
O significado biológico e patológico das diferentes isoformas de proteínas SNCA ainda não foi totalmente descoberto. Entretanto, isoformas específicas SNCA pós modificação translacional e emendas foram associadas a propensões de agregação intracelular (Kalivendi et al., 2010) e são expressas diferentemente em sinucleinopatias humanas (Beyer et al., 2008; Beyer e Ariza, 2012). Estudos de SNCA pós modificação translacional mostraram que os corpos de Lewy, a marca patológica das sinucleinopatias, contêm abundantes SNCA fosforiladas, nitradas e monoubiquitadas (Kim et al., 2014). Os efeitos da modificação pós-transcripcional na agregação de SNCA também têm sido estudados. Sugeriu-se emendas alternativas para afetar a agregação de SNCA. Uma deleção do exon 3 ou 5 prevê consequências funcionais: enquanto o exon 3 (SNCA126) leva à interrupção do domínio de interação proteína-membrana N-terminal que pode levar a menos agregação, e o exon 5 (SNCA112) pode resultar em maior agregação devido a um encurtamento significativo do C-terminus não estruturado (Lee et al., 2001; Beyer, 2006). No córtex frontal da DLB, o SNCA112 aumenta acentuadamente em relação aos controles (Beyer et al., 2008), enquanto os níveis de SNCA126 diminuem no córtex pré-frontal dos pacientes com DLB (Beyer et al., 2006). Em contraste, a expressão SNCA126 mostrou aumento no córtex frontal do cérebro da DP e nenhuma diferença significativa na AEM (Beyer et al., 2008). SNCA98 é uma variante de emenda específica do cérebro que carece de exon 3 e 5 e exibe diferentes níveis de expressão em várias áreas do cérebro fetal e adulto. A superexpressão do SNCA98 foi relatada em DLB, PD (Beyer et al., 2007) e MSA (Beyer et al., 2008), corticais frontais comparadas com controles. Além disso, o processo pós transcrição resultando em uso alternativo 3′UTR foi relatado para ter efeitos na estabilidade e localização do mRNA (Fabian et al., 2010; Rhinn et al., 2012; Yeh e Yong, 2016). Investigações adicionais sobre as propensões de agregação das diferentes isoformas conhecidas da proteína SNCA e a composição dos corpos de Lewy são justificadas. Além disso, nosso estudo estabeleceu as bases para análises de quantificação de mRNA das transcrições previamente conhecidas e novas em uma amostra maior composta de sujeitos com uma gama de estágios clinicopatológicos usando várias regiões cerebrais de cada sujeito. Estas análises da paisagem transcriptômica específica da região cerebral de SNCA no contexto da gravidade neuropatológica serão informativas com relação ao papel de isoformas específicas de transcrição de SNCA na progressão dos estágios neuropatológicos e da gravidade dos corpos de Lewy e da densidade das neurites Lewy.
Neste trabalho, focamos na criação de um padrão de seqüenciamento e análise para análise de dados de gDNA e cDNA direcionados, gerados a partir dos mesmos sujeitos. Esta é uma abordagem poderosa que potencialmente permite o faseamento das seqüências de gDNA através da região completa de um gene em particular com base na heterozigosidade na seqüência das isoformas de transcrição de comprimento total. Os dados de gDNA do PacBio neste estudo produziram blocos faseados que cobriram 81% da região de 114 kb centrada na SNCA, com o bloco faseado mais longo excedendo 54 kb. Como a faseação do gDNA é limitada pelo comprimento de leitura e heterozigosidade, o aumento do comprimento de leitura provavelmente gerará blocos de fase maiores.
gDNA análise de variantes confirmadas, conhecidas e identificadas novas repetições curtas em tandem (STRs) nas regiões intrônicas. Por exemplo, anteriormente, usando sequenciamento de fases por clonagem e sequenciamento Sanger, descobrimos quatro haplótipos distintos dentro de uma região rica em CT intrônica que compreendia um cluster de sequências repetitivas variáveis (Lutz et al., 2015). Mostramos que um haplótipo específico, denominado haplótipo 3, conferia risco para desenvolver a patologia corporal de Lewy em pacientes com Alzheimer. Aqui, validamos a sequência desta região altamente polimórfica de baixa complexidade e os seus quatro haplótipos definidos. Embora nosso tamanho da amostra fosse pequeno, o “haplótipo 3” estava presente exclusivamente em pacientes com doença (um paciente DP, dois pacientes DLB), consistente com nossos achados anteriores. Os resultados piloto e a nossa publicação anterior fornecem a premissa de repetir as análises de associação de sinucleinopatias com sinucleopatias definidas com precisão, ou seja, por leituras longas, STRs e haplótipos estruturais utilizando um tamanho de amostra maior.
O nosso artigo demonstrou a capacidade do sistema PacBio Sequel em descobrir transcrições completas de um gene implicado numa doença e caracterizar o repertório completo de transcrições completas de um gene implicado numa doença. Além disso, também mostramos que o gDNA de leitura longa define com mais precisão variantes estruturais curtas e haplótipos incluindo STRs e por isso pode facilitar a descoberta e validação de variantes associadas à doença outras que os SNPs. Coletivamente, este novo conhecimento é altamente valioso e aplicável no avanço do nosso entendimento das etiologias genéticas, que podem envolver transtornos no cenário transcript, doenças humanas complexas subjacentes, incluindo doenças neurodegenerativas relacionadas à idade, como sinucleinopatias.
Dados Disponibilidade
As três células SMRT dos dados brutos do gDNA estão disponíveis em Zenodo.org com doi: 10.5281/zenodo.1560688. A única célula SMRT de dados crus de cDNA está disponível em Zenodo.org com doi: 10.5281/zenodo.1581809. Os resultados processados de gDNA e cDNA, incluindo as variantes de gDNA e isoformas de cDNA, estão disponíveis em Zenodo.org com doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F contribuiu com a concepção e design do estudo. ET e WR organizaram bases de dados de seqüências, realizaram as análises de seqüenciamento e prepararam todas as figuras e tabelas. O-CG e JB manipularam os tecidos cerebrais e prepararam as amostras nucleicas. TH gerou os conjuntos de dados de seqüenciamento. A SK desenhou e obteve os reagentes. OC-F, ET e WR escreveram o primeiro rascunho do manuscrito. OC-F obteve o financiamento. Todos os autores contribuíram para a preparação do manuscrito, leram e aprovaram a versão submetida.
Funding
Este trabalho foi financiado em parte pelo National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Conflict of Interest Statement
ET, WR, TH, e SK são ou eram funcionários da Pacific Biosciences no momento do estudo.
Os autores restantes declaram que a pesquisa foi realizada na ausência de quaisquer relações comerciais ou financeiras que pudessem ser interpretadas como um potencial conflito de interesses.
Agradecimentos
Este manuscrito foi lançado como pré-impressão na BioRxiv (Tseng et al., 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Material Suplementar
O Material Suplementar para este artigo pode ser encontrado online em: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona study of aging and neurodegenerative disorders and brain and body donation program. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-estrutura da sinucleína, modificação pós-tradução e emendas alternativas como melhoradores de agregação. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., e Ariza, A. (2012). Modificação alfa-sinucleína pós-tradução e emendas alternativas como gatilho para neurodegeneração. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., e Ariza, A. (2008). Expressão diferencial de isoformas alfa-sinucleína, parkin e sinfilina-1 na doença corporal de Lewy. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., e Ariza, A. (2007). Identificação e caracterização de uma nova isoforma alfa-sinucleína e seu papel nas doenças do corpo de Lewy. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Baixos níveis de alfa-sinucleína 126 mRNA em demência com corpos de Lewy e doença de Alzheimer. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., e Filipowicz, W. (2010). Regulação da tradução e estabilidade do mRNA por microRNAs. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: alinhamento de sequências múltiplas com alta precisão e alto rendimento. Ácidos nucléicos Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., e Kalyanaraman, B. (2010). Os oxidantes induzem emendas alternativas de Α-synuclein: implicações para a doença de Parkinson. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., e Halliday, G. M. (2014). Biologia alfa-sinucleína nas doenças do corpo de Lewy. Res. de Alzheimer. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). A variante receptora de androgênio AR-V9 é coexpressa com AR-V7 em metástases do câncer de próstata e prevê a resistência à abiraterona. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). A membrana Α-synuclein tem uma alta propensão de agregação e a capacidade de semear a agregação da forma citosólica. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., e Chiba-Falek, O. (2015). Um haplótipo rico em citosina-timina (CT) no intron 4 da SNCA confere risco para a patologia corporal de Lewy na doença de Alzheimer e afeta a expressão da SNCA. Alzheimer Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: phasing rápido e preciso baseado em leitura. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., e Perry, R. H. (1999). Relatório do segundo workshop internacional sobre demência com Lewy Body: diagnóstico e tratamento. Consórcio sobre demência com corpos de Lewy. Neurologia 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., e Isacson, O. (2012). Níveis de expressão transcritos da alfa-sinucleína de comprimento total e suas três variantes alternadamente emendadas nas regiões cerebrais da doença de Parkinson e em um modelo transgênico de sobreexpressão da alfa-sinucleína em camundongos. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Escala da descoberta da variante genética precisa para dezenas de milhares de amostras. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternativa Α – uso da transcrição da sinucleína como mecanismo convergente na patologia da doença de Parkinson. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10,1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., e Südhof, T. C. (2014). Cartografia de emendas alternativas de neurexina mapeadas por sequenciamento de mRNA de leitura longa de uma única molécula. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long read sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). Expressão alterada da paisagem das variantes de emenda FMR1 em portadores de pré-mutação. Biochim. Biófilos. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., e Yu, J.-T. (2014). A ligação entre o gene SNCA e o Parkinsonismo. Neurobiol. Envelhecimento 36, 1-14. doi: 10.1016/j.neurobiolage.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., e Yong, J. (2016). Poladenilação alternativa de mRNAs: 3′- a região não traduzida importa na expressão gênica. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar