- Introducere
- Materiale și metode
- Eșantioane de studiu
- Extracții de ADN genomic și ARN
- Pregătirea bibliotecii și secvențierea
- Capturarea ADNg cu ajutorul sondelor IDT Xgen® Lockdown® și secvențierea unei singure molecule
- Capturarea ADN-ului utilizând sondele IDT Xgen® Lockdown® și secvențierea izoformelor cu o singură moleculă (Iso-Seq)
- Analiză ADNg
- Short Variant Analysis and Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Isoform Analysis
- Isoform SNP Calling
- Rezultate
- Capturarea țintită a gADN-ului a identificat variații cunoscute și noi
- Targeted cDNA Capture Identified Novel Start and End Sites
- Full-Length cDNA Enables Isoform-Level Phasing Information
- Discuție
- Disponibilitatea datelor
- Contribuții ale autorilor
- Finanțare
- Declarație privind conflictul de interese
- Acknowledgments
- Supplementary Material
Introducere
Programele transcripționale și posttranscripționale controlează nivelurile de expresie a genelor și/sau producția de mai multe izoforme distincte de ARNm, iar modificările acestor mecanisme au ca rezultat dereglarea expresiei genelor și profile de expresie diferențială. Reglarea genetică transcripțională și posttranscripțională aberantă este abundentă în țesuturile sistemului nervos uman și contribuie la diferențele fenotipice în cadrul indivizilor și între indivizi, în stare de sănătate și boală.
Disreglarea expresiei alfa-sinucleinei a fost implicată în patogeneza sinucleinopatiilor, în special a bolii Parkinson (PD) și a demenței cu corpi Lewy și (DLB). În timp ce rolul supraexpresiei SNCA în sinucleopatii, în special în PD, a fost bine stabilit, aici ne-am concentrat pe determinarea repertoriului complet al izoformelor transcrise ale SNCA în diferite sinucleopatii. Anterior, au fost descrise mai multe izoforme diferite ale transcripției SNCA pentru gena SNCA, apărute în urma splicingului alternativ, a situsurilor de început transcripțional (TSS) și a selecției situsurilor de poliadenilare (McLean et al., 2012; Xu et al., 2014). Splicarea alternativă a exonilor codificatori dă naștere la SNCA 140, SNCA 112, SNCA 126 și SNCA 98, rezultând patru izoforme proteice (Beyer și Ariza, 2012). TSS-urile alternative ale genei SNCA au ca rezultat patru 5′UTR-uri diferite, iar selecția alternativă a diferitelor situsuri de poliadenilare determină trei lungimi majore ale 3′UTR, fără impact asupra compoziției produsului proteic (Beyer și Ariza, 2012). Obiectivul nostru general este de a obține noi informații despre contribuția diferitelor specii de ARNm SNCA, cunoscute și noi, la patogeneza și eterogenitatea sinucleinopatiilor.
Până în prezent, majoritatea studiilor au utilizat tehnologii de secvențiere cu citire scurtă pentru a interoga complexitatea transcriptomului în creierele umane. Disponibilitatea tehnologiilor de citire lungă de a treia generație oferă o imagine fără precedent și aproape completă a structurilor izoformelor. Cu toate acestea, secvențierea existentă a transcrierilor cu citire lungă pentru genele bolilor umane a utilizat o abordare bazată pe ampliconi (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Deși această abordare a avut succes în identificarea splicingului alternativ complex în genele bolilor umane, ea este limitată la proiectarea amorselor PCR și nu va descoperi situsurile alternative de început și sfârșit. Îmbogățirea țintită, cum ar fi prin utilizarea sondelor IDT, poate oferi o vizualizare cuprinzătoare a izoformelor genelor de interes la un cost redus de secvențiere. Mai mult, citirile foarte precise ale transcripției complete permit haplotiparea specifică a izoformei.
În acest studiu, prezentăm primul studiu cunoscut care utilizează capturarea țintită a ADNg și ADNc din regiunea genei SNCA folosind secvențierea PacBio SMRT. Regiunea genei SNCA are o lungime de ~114 kb, constând din șase exoni cu lungimi de transcriere de aproximativ 3 kb. Am multiplexat 12 probe de creier uman din PD, DLB și probe de control normale și am secvențiat biblioteca de ADNg și ADNc pe sistemul PacBio Sequel. Descriem analizele bioinformatice utilizate pentru a identifica SNP-uri, indels și repetări scurte în tandem pentru captura ADNg și haplotiparea la nivel de izoformă pentru datele ADNc. Arătăm că captura țintită este o modalitate rentabilă de a studia în comun variația genomică și splicingul alternativ într-o genă neuronală legată de o boală.
Materiale și metode
Eșantioane de studiu
Cohorta de studiu (N = 12) a constat din persoane cu trei diagnostice neuropatologice confirmate prin autopsie: (1) PD (N = 4); (2) DLB (N = 4); și (3) subiecți normali din punct de vedere clinic și neuropatologic (N = 4). Țesuturile creierului cortexului frontal au fost obținute prin intermediul Kathleen Price Bryan Brain Bank (KPBBB) de la Universitatea Duke, Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) și Layton Aging and Alzheimer’s Disease Center de la Oregon Health and Science University. Fenotipurile neuropatologice au fost determinate în urma examinării postmortem după metode standard bine stabilite, urmând metoda și recomandările de practică clinică ale lui McKeith și ale colegilor (McKeith et al., 1999, 2005). Densitatea patologiei LB (într-un set standard de regiuni cerebrale) a primit scoruri de ușoară, moderată, severă și foarte severă. Eșantioanele de studiu din cadrul fiecărui grup de diagnostic, PD și DLB, au fost atent selectate astfel încât severitatea fenotipurilor clinicopatologice să fie similară în cadrul fiecărei patologii. Toate creierele au prezentat corpuri Lewy (LB) în trunchiul cerebral, limbic și neocortical, în timp ce PD a prezentat scoruri McKeith severe până la foarte severe în subnigra și amigdala. Toate creierele indică faptul că nu există DA în conformitate cu criteriile CERAD și cu stadiul Braak și Braak = II. Probele de creier sănătos din punct de vedere neurologic au fost obținute din țesuturi post-mortem ale unor subiecți normali din punct de vedere clinic, care au fost examinați, în cele mai multe cazuri, în termen de 1 an de la deces și s-a constatat că nu prezentau tulburări cognitive sau parkinsonism și constatări neuropatologice insuficiente pentru diagnosticarea PD, a bolii Alzheimer (AD) sau a altor tulburări neurodegenerative. Toate eșantioanele au fost albi. Datele demografice și neuropatologia pentru acești subiecți sunt rezumate în Tabelul suplimentar 1. Proiectul a fost aprobat de către Duke Institution Review Board (IRB), care a oferit o aprobare etică. Metodele au fost efectuate în conformitate cu liniile directoare și reglementările relevante.
Extracții de ADN genomic și ARN
DNA genomic a fost extras din țesuturile cerebrale prin protocolul standard Qiagen (Qiagen, Valencia, CA). ARN total a fost extras din probele de creier (100 mg) folosind reactivul TRIzol (Invitrogen, Carlsbad, CA), urmat de purificare cu un kit RNeasy (Qiagen, Valencia, CA), conform protocolului producătorului. concentrația de ADNg și ARN a fost determinată spectrofotometric, iar calitatea probelor de ARN și lipsa degradării semnificative au fost confirmate prin măsurători ale numărului de integritate al ARN (RIN, tabelul suplimentar 1) utilizând un bioanalizator Agilent.
Pregătirea bibliotecii și secvențierea
Capturarea ADNg cu ajutorul sondelor IDT Xgen® Lockdown® și secvențierea unei singure molecule
Aproximativ 2 μg din fiecare probă de ADNg a fost secționată la 6 kb cu ajutorul Covaris g-TUBE și ligaturată cu adaptoare cu cod de bare. Un grup echimolar de bibliotecă de ADNg cu 12 complexe cu cod de bare (2 μg în total) a fost introdus în captura bazată pe sondă cu un panou de gene SNCA proiectat la comandă.
A fost construită o bibliotecă SMRTBell folosind 626 ng de ADNg capturat și re-amplificat1.
Capturarea ADN-ului utilizând sondele IDT Xgen® Lockdown® și secvențierea izoformelor cu o singură moleculă (Iso-Seq)
Aproximativ 100-150 ng de ARN total pe reacție a fost transcris în sens invers utilizând kitul de sinteză cADN Clontech SMARTer și 12 oligo dT cu cod de bare specifice probei (cu secvențe de cod de bare PacBio 16mer, a se vedea Metode suplimentare). Trei reacții de transcriere inversă (RT) au fost procesate în paralel pentru fiecare eșantion. Optimizarea PCR a fost utilizată pentru a determina numărul optim de cicluri de amplificare pentru reacțiile PCR la scară largă din aval. Un singur primer (primer IIA din kitul Clontech SMARTer 5′ AAG CAG TGG TGG TAT CAA CGC AGA GTA C 3′) a fost utilizat pentru toate reacțiile PCR post-RT. Produsele PCR la scară mare au fost purificate separat cu margele 1X AMPure PB, iar bioanalizatorul a fost utilizat pentru QC. Un grup echimolar de bibliotecă de ADNc cu cod de bare 12-plex (1 μg în total) a fost introdus în captura pe bază de sondă cu un panou de gene SNCA proiectat la comandă.
A fost construită o bibliotecă SMRTBell folosind 874 ng de ADNc capturat și re-amplificat2. O celulă SMRT Cell 1M (film de 6 h) a fost secvențiată pe platforma PacBio Sequel folosind chimia 2.0.
Analiză ADNg
Secvențierea datelor ADNg cu cod de bare a fost efectuată pe trei celule SMRT Cell 1M folosind chimia 2.0. Datele au fost demultiplexate prin rularea aplicației Demultiplex Barcodes în PacBio SMRT Link v6.0.
Short Variant Analysis and Phasing
Circular Consensus Sequence (CCS) reads au fost generate folosind SMRT Analysis 6.0 din fiecare set de date demultiplexate și aliniate la genomul de referință hg19 folosind minimap2. Dublurile PCR din amplificarea post-captură au fost identificate prin cartografierea punctelor finale și etichetate cu ajutorul unui script personalizat. Variantele scurte au fost apelate utilizând GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). După o primă trecere de filtrare utilizând profunzimea de acoperire și metricele de calitate, variantele au fost inspectate manual în IGV3. În cazul în care variantele nu se aflau în fază cu SNP-urile din apropiere, acestea au fost filtrate manual. Locurile variantelor care au trecut de curatoria manuală au fost utilizate împreună cu alinierile CCS deduplicate pentru punerea în fază cu read-backed cu WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Sub-secvențele aliniate la chr4: 90742331-90742559 (hg19) au fost extrase pentru fiecare eșantion. După ce s-a inspectat distribuția dimensională a acestor subsecvențe, acestea au fost grupate în funcție de mărimea și similaritatea secvenței folosind o combinație de python și MUSCLE (Edgar, 2004), iar o secvență consensuală a fost generată independent pentru fiecare grup.
Scripturi personalizate și fluxuri de lucru descrise în continuare în https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Isoform Analysis
Secvențierea datelor ADNc cu coduri de bare s-a făcut pe un SMRT Cell 1M pe sistemul PacBio Sequel folosind chimia 2.0. Analiza bioinformatică a fost efectuată cu ajutorul aplicației IsoSeq3 din PacBio SMRT Analysis v6.0.0.0 pentru a obține secvențe de izoforme de înaltă calitate, de lungime completă (a se vedea Metode suplimentare pentru mai multe informații).
Isoform SNP Calling
Lecturile de lungime completă asociate cu cele 41 de izoforme finale din toate cele 12 probe au fost aliniate la genomul hg19 pentru a crea un pileup. Bazele cu QV mai mic de 13 au fost excluse. Apoi, la fiecare poziție cu o acoperire de cel puțin 40 de baze, s-a aplicat un test Fisher exact cu corecția Bonferroni cu un prag p de 0,01. Au fost apelate numai SNP de substituție care nu se află în apropierea regiunilor homopolimerice (întinderi de 4 sau mai multe nucleotide identice). După apelarea SNP, genotipul pentru fiecare eșantion a fost determinat prin numărarea numărului de citiri de lungime completă (FL) specifice eșantionului. În cazul în care un eșantion avea 5+ citiri FL care susțineau atât baza de referință, cât și baza alternativă, acesta era heterozigot. În cazul în care un eșantion avea 5+ citiri FL care susțineau o alelă și mai puțin de 5 citiri FL pentru cealaltă, acesta era homozigot. În caz contrar, a fost neconcludent. Scripturile sunt disponibile la: https://github.com/Magdoll/cDNA_Cupcake.
Rezultate
Am conceput sonde personalizate pentru gena SNCA și am efectuat o captură țintită atât a ADNg, cât și a ADNc pe o bibliotecă multiplexată formată din 12 probe de creier uman din PD, DLB și controale normale (Figura 1, Tabelul suplimentar 1). Bibliotecile de gADN și ADNc au fost secvențiate pe platforma PacBio Sequel. Analiza bioinformatică a fost efectuată cu ajutorul software-ului PacBio, urmată de o analiză personalizată.
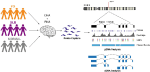
Figura 1. Prezentarea schematică a designului studiului. Materialele ADN și ARN au fost extrase din țesuturile cerebrale postmortem ale pacienților cu boala Parkinson, demență cu corpuri Lewy și din grupurile de control. bibliotecile de ADNg și ADNc au fost realizate prin hibridare cu sonde și secvențiate pe sistemul PacBio Sequel. Analiza a fost efectuată cu ajutorul software-ului PacBio și a altor instrumente existente.
Capturarea țintită a gADN-ului a identificat variații cunoscute și noi
După generarea secvențelor consensuale circulare (CCS) și eliminarea duplicatelor PCR (Metode suplimentare), am obținut o acoperire unică medie de 16 până la 71 de ori a regiunii genei SNCA. Lecturile CCS au avut o lungime medie de inserție de 2,9 kb și o precizie medie de citire de 98,9%. Cu excepția unei regiuni de 5 kb neacoperite în mod intenționat de sonde datorită prezenței elementelor LINE (hg19 chr4: 90697216-90702113) și a unei regiuni de 2,1 kb cu conținut ridicat de GC în jurul exonului 1, a existat o acoperire suficientă pentru a genotipa ambele haplotipuri pentru fiecare dintre cele 212 probe (Figura 2, Figura suplimentară 1).

Figura 2. Capturarea și punerea în fază a gADN-ului țintit. Un exemplu care prezintă un eșantion din fiecare condiție. Pista de sus arată una dintre izoformele SNCA, urmată de acoperirea gDNA pentru cele trei probe. Pista variantelor arată fiecare SNP și sunt codificate prin culoare pentru heterozigot (violet), homozigot alternativ (portocaliu) și homozigot de referință (gri). Blocurile fazate sunt afișate în albastru deschis. Pista de jos arată locațiile sondei de captură. Regiunea de abandon în proiectarea sondei se datorează celor două elemente LINE din mijlocul intronului 4. Pentru informații privind acoperirea gDNA și punerea în fază a tuturor celor 12 probe, a se vedea figurile suplimentare.
Utilizând GATK4 HC, filtrarea bazată pe calitate și curatoria manuală, am identificat 282 de SNP și 35 de indeluri, inclusiv 8 SNP și 13 indeluri care nu se găsesc în dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (tabelul suplimentar 2). Nu a fost identificată nicio variantă în regiunea de codificare pentru SNCA, deși au fost identificate opt variante în regiunile netranslate. Majoritatea variantelor identificate, inclusiv mai multe repetări scurte în tandem (STR), se încadrează în intronii 2, 3 și 4.
Am descris anterior o regiune bogată în CT extrem de polimorfică în intronul 4 al SNCA cu patru haplotipuri observate (Lutz et al., 2015). Deși această regiune foarte repetitivă și structural variabilă s-a dovedit a fi dificil de genotipat cu GATK4 HC, am reușit să construim secvențe consensuale pentru toate cele 12 probe și am observat toate cele 4 haplotipuri descoperite anterior (Figura suplimentară 2). În plus, am identificat un nou STR în intronul 4, care constă într-o unitate de trei baze repetată de 16 ori în referință. În cadrul celor 12 probe, am identificat trei haplotipuri, cu 9, 12 și 15 copii ale unității repetate TTG. GATK HC a genotipat corect toate acestea, cu excepția unui haplotip pentru PD-4, care a avut o acoperire destul de scăzută în această regiune. Cu toate acestea, cu datele date pentru acest eșantion, genotipul poate fi determinat prin inspecție vizuală (tabelul 1).

Tabel 1. O nouă repetiție în tandem de triplet în intronul 4 (chr4: 90713442).
Am folosit variantele scurte detectate de GATK HC împreună cu instrumentul de fazare bazat pe citire WhatsHap (Martin et al., 2016) pentru a face o fază a citirilor CCS de-a lungul locusului, cu o gamă de succes determinată în principal de densitatea variantelor heterozigote de-a lungul locusului. Probele PD-1, PD-4, N-4, DLB-1 și DLB-4 au avut întinderi lungi de heterozigozitate scăzută, cu foarte puține blocuri de fază scurte, în timp ce celelalte probe au produs blocuri de fază variind de la 7 la 18 ori lungimea medie de citire, până la 54 kb (Figura suplimentară 3).
Targeted cDNA Capture Identified Novel Start and End Sites
Am procesat datele PacBio cDNA (Iso-Seq) utilizând software-ul PacBio SMRT Analysis. După cartografierea datelor Iso-Seq la hg19 și eliminarea artefactelor (tabelul suplimentar 3, figura suplimentară 4), am obținut un set final de 41 de izoforme SNCA (figura 3). Toate izoformele finale au toate situsurile de îmbinare canonice (GT-AG sau GC-AG) și sunt susținute de un total de 20 sau mai multe lecturi de lungime completă. Majoritatea izoformelor (28 din 41) au toți cei șase exoni, diferind doar prin utilizarea unor situsuri de început 5′ alternative și lungimi 3′ UTR. Lungimile 3′ UTR au variat între 300 și 2,6 kb. Utilizarea unui situs de pornire 5′ alternativ foarte divers în SNCA este cunoscută; ceea ce este mai puțin cunoscut este lungimea variabilă a 3′ UTR, care a fost studiată anterior folosind date RNA-seq care nu au rezolvat structurile izoformei de lungime completă (Rhinn et al., 2012). Datele Iso-Seq arată că lungimea variabilă 3′ UTR pare să fie împerecheată cu toate combinațiile posibile de situsuri de pornire 5′, fără o cuplare preferențială. Aproape niciuna dintre variabilitățile situsului de început și de sfârșit nu modifică cadrul de citire deschis prezis (Figura suplimentară 5) și se preconizează că se va traduce în secvența canonică de 141 de aminoacizi.

Figura 3. Izoformele SNCA capturate cu ajutorul Iso-Seq-ului țintit identifică noi situsuri de început și sfârșit. Cea mai mare parte a complexității izoformelor provine din utilizarea combinatorie a unor lungimi alternative ale 3′ UTR și a exonului 1, cu câteva situsuri alternative rare de îmbinare găsite în exonul 1 (verde), 2 (roșu) și 4 (albastru). Toate joncțiunile au situsuri de racordare canonice. Am identificat cinci izoforme care au omis exonul 5 și două izoforme care au omis exonul 3. De asemenea, am identificat noi situsuri de început (portocaliu) și de sfârșit (violet) în intronul 4. SNP-urile apelate sunt marcate în violet.
Am validat în continuare joncțiunile noi (dar canonice) folosind date de joncțiune cu citire scurtă disponibile în mod public. Baza de date Intropolis (v1, https://github.com/nellore/intropolis) combină peste 21.000 de date RNA-seq disponibile public. Din cauza volumului mare de date de joncțiune susținute de o singură citire scurtă, pentru acest studiu, avem nevoie de un minim de 10 suporturi de citire scurtă (combinate din toate >21.000 de seturi de date RNA-seq) pentru a confirma joncțiunile noastre noi Iso-Seq. Cu excepția joncțiunilor noi pentru PB.1016.253 și PB.1016.296 (Figura 3), toate celelalte joncțiuni noi sunt susținute de setul de date Intropolis. Este interesant faptul că aceste joncțiuni noi au o susținere a lecturilor scurte semnificativ mai mică decât joncțiunile anunțate de Gencode. De exemplu, cele două joncțiuni noi din PB.1016.139 introduse de exonul nou sunt susținute de 2 519 și, respectiv, 44 de citiri scurte Intropolis, în timp ce celelalte patru joncțiuni cunoscute sunt susținute de peste 1 milion de citiri scurte. Acest lucru arată puterea îmbogățirii țintite folosind secvențierea transcriptomului pe toată lungimea pentru detectarea izoformelor rare, noi.
Am observat două izoforme cu omiterea exonului 3 (SNCA126) și cinci izoforme cu omiterea exonului 5 (SNCA112). Din nou, diversitatea de splicing în aceste două grupuri de omitere a exonului provine în principal din utilizarea diversă a situsurilor de pornire 5′ alternative și din lungimea variabilă a 3′ UTR. Predicția ORF arată că omiterea exonului 3 sau a exonului 5 scurtează ORF-ul, dar menține cadrul de citire. Trei izoforme au noi situsuri de capăt 3′ situate în intronul 4. Predicția ORF arată că acest lucru are ca rezultat un produs proteic trunchiat.
Am identificat un situs de început 5′ neanunțat anterior, situat în intronul 4 (hg19 chr4: 90692548-90693045, figura 3). Cele trei izoforme asociate cu acest nou start constau din noul sit de start, exonul 5 și lungimi variabile de 3′ UTR. În mod interesant, în timp ce datele de citire scurtă descărcate public de la GTEx și Sandor et al. (2017) și datele de vârf CAGE (FANTOM5) nu au susținut acest nou sit de început, un set de date publice recente NA12878 direct RNA4 conținea doar o singură transcripție SNCA care a confirmat acest sit de început alternativ. Mai mult, joncțiunea nouă dintre exonul 5 și noul site de pornire este confirmată de datele de joncțiune cu citire scurtă Intropolis. În mod interesant, se preconizează că acest nou sit de pornire 5′ introduce noi peptide, menținând în același timp cadrul de citire în exonul 5.
Am identificat, de asemenea, trei transcripte SNCA cu noi situsuri finale (Figura 3). Două izoforme (PB.1016.383, PB.1016.384) au utilizat o UTR 3′ extinsă în exonul 4, în timp ce a treia izoformă (PB.1016.381) a utilizat un nou exon 3′ în intronul 4. Joncțiunile noi dintre ultimul exon nou și exonul anterior sunt susținute de datele publice privind joncțiunile de citire scurtă (Intropolis). Noile 3′ UTR-uri noi au ca rezultat o predicție ORF trunchiată.
Utilizând numărul normalizat de citiri pe toată lungimea ca indicator pentru abundența izoformei, am constatat că una dintre izoformele SNCA canonice (PB.1016.131) este cea mai abundentă, cu o abundență de 50-60% în toate probele subiectului (tabelul suplimentar 4). Am grupat în continuare cele 41 de izoforme în funcție de modelele lor de splicing (Tabelul 2). Izoformele care au toți cei șase exoni reprezintă 95-97% din abundență. Studiile anterioare au arătat o creștere marcată a expresiei izoformelor cărora le lipsește exonul 3 (SNCA126) în cortexul frontal al eșantioanelor DLB în comparație cu cele normale (Beyer et al., 2008); numărătoarea noastră agregată a izoformelor arată că trei dintre eșantioanele DLB au un nivel de numărare ușor ridicat în comparație cu eșantioanele normale, precum și variantele SNCA112 (omiterea exonului 5) pentru PD și DLB față de eșantioanele normale.

Tabel 2. Abundența izoformei SNCA pentru fiecare probă, agregată în funcție de modelele de splicing.
Full-Length cDNA Enables Isoform-Level Phasing Information
Am apelat SNP-uri folosind cDNA prin acumularea tuturor citirilor de lungime completă din cele 12 probe pentru a apela variantele (a se vedea secțiunea “Metode”). În total, au fost apelate patru SNP-uri și toate au fost adnotate anterior în dbSNP (Tabelul 3, Figura 3). Cele patru SNP-uri sunt toate localizate în regiuni non-CDS, unul în 3′ UTR (exonul 6), unul în intronul 4 și două în 5′ UTR (exonul 1). SNP 3′ UTR (chr4: 90646886) este acoperit doar de izoformele cu o UTR 3′ care are o lungime de cel puțin ~1 kb și, prin urmare, nu toate izoformele canonice acoperă acest SNP. SNP-ul intronului 4 (chr4: 90743331) este acoperit doar de noile izoforme alternative de capăt 3′ (PB.1016.383, PB.1016.384) și nu este conectat la niciunul dintre celelalte SNP-uri. Cele două SNP 5′ UTR (chr4: 90757312 și chr4: 90758389) sunt acoperite de două utilizări ale exonului 1 care se exclud reciproc și, prin urmare, nu sunt, de asemenea, legate.

Tabelul 3. Informații SNP ADNc.
Abordarea noastră actuală este limitată la apelarea numai a variantelor de substituție în regiunile transcrise cu o acoperire suficientă. Compararea listei SNP-urilor noastre cu adnotarea hg19 dbSNP arată că majoritatea SNP-urilor sau variantelor ratate fie aveau o frecvență mai mică de 1% în populație, fie nu erau substituții de un singur nucleotid, fie erau adiacente unor regiuni de complexitate redusă. De exemplu, rs77964369 (chr4: 90646532) este raportat ca având o frecvență 50/50 de T/A; cu toate acestea, acest T este adiacent la o porțiune de 11 genom As în aval. Inspecția manuală a pilei de citire Iso-Seq, care are ~1.300 de citiri la acest sit, nu sugerează dovezi de variație, cel puțin între cele 12 eșantioane ale noastre.
Utilizând citirile specifice eșantioanelor, numim genotipul fiecărui eșantion la fiecare locație SNP (Tabelul 3). În afară de PD-2 care are prea puține citiri și nu este concludent pentru toate cele patru SNP-uri, am reușit să apelăm genotipul pentru majoritatea celorlalte probe. În special, DLB-3 a fost singurul eșantion care este heterozigot la toate locațiile SNP. În rest, nu am observat niciun model specific afecțiunii de preferință a unui genotip în detrimentul altuia.
Discuție
Descriem primul studiu care utilizează îmbogățirea țintită a genei SNCA pe biblioteci multiplexate de ADNg și ADNc pentru studierea bolilor neurologice folosind secvențierea cu citire lungă. Lungimile lungi de citire ale sistemului PacBio Sequel au facilitat secvențierea întregului repertoriu de izoforme de transcripție a genei SNCA. Am evidențiat diversitatea în ceea ce privește utilizarea situsurilor de pornire 5′ alternative și a lungimilor variabile ale 3′ UTR și am observat evenimente cunoscute de omitere a exonilor, cum ar fi deleția exonului 3 (SNCA126) și deleția exonului 5 (SNCA112). În plus, au fost identificate noi situsuri alternative de început și sfârșit în cadrul intronului mare 4, care se preconizează că vor fi traduse în proteine noi. Este probabil că profunzimea ridicată a acoperirii de secvențiere a capturii țintite, în combinație cu capacitatea de a secvenția transcripții complete, ne-a permis să detectăm aceste izoforme nedescrise anterior.
Semnificația biologică și patologică a diferitelor izoforme ale proteinei SNCA nu a fost încă descoperită pe deplin. Cu toate acestea, izoformele specifice de modificare posttranslațională și de splicing ale SNCA au fost asociate cu propensiuni de agregare intracelulară (Kalivendi et al., 2010) și sunt exprimate diferit în sinucleopatiile umane (Beyer et al., 2008; Beyer și Ariza, 2012). Studiile privind modificarea post-traducțională a SNCA au arătat că corpurile Lewy, semnul patologic distinctiv al sinucleinopatiilor, conțin SNCA fosforilat, nitrat și monobiquitinat abundent (Kim et al., 2014). Efectele modificării post-transcripționale asupra agregării SNCA au fost, de asemenea, studiate. S-a sugerat că splicingul alternativ afectează agregarea SNCA. O deleție fie a exonului 3, fie a exonului 5 prezice consecințe funcționale: în timp ce deleția exonului 3 (SNCA126) duce la întreruperea domeniului N-terminal de interacțiune proteină-membrană, ceea ce poate duce la o agregare mai mică, iar deleția exonului 5 (SNCA112) poate duce la o agregare sporită datorită unei scurtări semnificative a părții C-terminale nestructurate (Lee et al., 2001; Beyer, 2006). În cortexul frontal al pacienților cu DLB, SNCA112 este crescut semnificativ în comparație cu controalele (Beyer et al., 2008), în timp ce nivelurile SNCA126 sunt scăzute în cortexul prefrontal al pacienților cu DLB (Beyer et al., 2006). În schimb, expresia SNCA126 a arătat o creștere a expresiei SNCA126 în cortexul frontal al creierelor PD și nu există diferențe semnificative în MSA (Beyer et al., 2008). SNCA98 este o variantă de racordare specifică creierului care este lipsită atât de exonul 3, cât și de exonul 5 și prezintă niveluri diferite de expresie în diferite zone ale creierului fetal și adult. Supraexpresia SNCA98 a fost raportată în cortexul frontal al DLB, PD (Beyer et al., 2007) și MSA (Beyer et al., 2008) în comparație cu controalele. În plus, s-a raportat că procesul post-transcripțional care duce la utilizarea alternativă a 3′UTR a avut efecte asupra stabilității și localizării ARNm (Fabian et al., 2010; Rhinn et al., 2012; Yeh și Yong, 2016). Sunt justificate investigații suplimentare cu privire la propensiunile de agregare ale diferitelor izoforme cunoscute ale proteinei SNCA și la compoziția corpurilor Lewy. Mai mult decât atât, studiul nostru a pus bazele unor analize de cuantificare a ARNm a transcriptelor cunoscute anterior și a celor noi într-un eșantion de dimensiuni mai mari, alcătuit din subiecți cu o gamă de stadii clinico-patologice, folosind mai multe regiuni ale creierului de la fiecare subiect. Aceste analize ale peisajului transcriptomic specific regiunii cerebrale a SNCA în contextul severității neuropatologice vor fi informative în ceea ce privește rolul izoformelor specifice ale transcriptelor SNCA în progresia stadiilor neuropatologice și severitatea densității corpurilor Lewy și a neviților Lewy.
În această lucrare, ne-am concentrat pe crearea unui standard de secvențiere și analiză pentru analiza datelor de ADNg și ADNc direcționate generate de la aceiași subiecți. Aceasta este o abordare puternică care permite, în mod potențial, eșalonarea secvențelor de ADNg în întreaga regiune completă a unei anumite gene, pe baza heterozigozității în secvența izoformelor transcrise pe toată lungimea. Datele de ADNg direcționate PacBio din acest studiu au produs blocuri fazate care au acoperit 81% din regiunea de 114 kb centrată pe SNCA, cel mai lung bloc fazat depășind 54 kb. Deoarece fasonarea gDNA este limitată de lungimea de citire și de heterozigozitate, creșterea lungimilor de citire va genera probabil blocuri de fază mai mari.
Analiza variantelor gDNA a confirmat variantele cunoscute și a identificat noi repetiții scurte în tandem (STR) în regiunile intronice. De exemplu, anterior, utilizând secvențierea în fază prin clonare și secvențiere Sanger, am descoperit patru haplotipuri distincte în cadrul unei regiuni intronice bogate în CT care cuprindea un grup de secvențe repetitive variabile (Lutz et al., 2015). Am arătat că un haplotip specific, denumit haplotipul 3, a conferit riscul de a dezvolta patologia corpului Lewy la pacienții cu Alzheimer. Aici, am validat secvența acestei regiuni extrem de polimorfe de complexitate redusă și a celor patru haplotipuri definite ale sale. Deși dimensiunea eșantionului nostru a fost mică, “haplotipul 3” a fost prezent exclusiv la pacienții bolnavi (un pacient cu PD, doi pacienți cu DLB), în concordanță cu constatările noastre anterioare. Rezultatele pilot și publicația noastră anterioară oferă premisa de a repeta analizele de asociere a sinucleinopatiilor cu STR-uri și haplotipuri structurale definite cu acuratețe, adică prin lecturi lungi, utilizând un eșantion de dimensiuni mai mari.
Articolul nostru a demonstrat capacitatea sistemului PacBio Sequel de a descoperi noi transcripte de lungime completă și de a caracteriza repertoriul complet de transcripte de lungime completă al unei gene implicate într-o boală. În plus, am arătat, de asemenea, că gDNA cu citire lungă definește cu mai multă acuratețe variantele structurale scurte și haplotipurile, inclusiv STR-urile și, prin aceasta, poate facilita descoperirea și validarea variantelor asociate bolilor, altele decât SNP-urile. În mod colectiv, aceste noi cunoștințe sunt extrem de valoroase și aplicabile în avansarea înțelegerii noastre a etiologiilor genetice, care pot implica perturbări în peisajul transcripțiilor, care stau la baza bolilor umane complexe, inclusiv a tulburărilor neurodegenerative legate de vârstă, cum ar fi sinucleinopatiile.
Disponibilitatea datelor
Cele trei celule SMRT de date brute gDNA sunt disponibile la Zenodo.org cu doi: 10.5281/zenodo.1560688. O celulă SMRT de date brute cADN este disponibilă la Zenodo.org cu doi: 10.5281/zenodo.1581809. Rezultatele prelucrate ale gDNA și cDNA, inclusiv variantele gDNA și izoformele cDNA, sunt disponibile la Zenodo.org cu doi: 10.5281/zenodo.3261805.
Contribuții ale autorilor
OC-F a contribuit la conceperea și proiectarea studiului. ET și WR au organizat bazele de date cu secvențe, au efectuat analizele de secvențiere și au pregătit toate figurile și tabelele. O-CG și JB s-au ocupat de țesuturile cerebrale și de prepararea probelor nucleice. TH a generat seturile de date de secvențiere. SK a proiectat și a obținut reactivii. OC-F, ET și WR au redactat prima versiune a manuscrisului. OC-F a obținut finanțare. Toți autorii au contribuit la pregătirea manuscrisului, au citit și au aprobat versiunea trimisă.
Finanțare
Această lucrare a fost finanțată parțial de National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Declarație privind conflictul de interese
ET, WR, TH și SK sunt sau au fost angajați ai Pacific Biosciences în momentul studiului.
Ceilalți autori declară că cercetarea a fost efectuată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
Acknowledgments
Acest manuscris a fost publicat ca pre-print la BioRxiv (Tseng et al., 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Supplementary Material
Materialul suplimentar pentru acest articol poate fi găsit online la adresa: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Studiul din Arizona privind îmbătrânirea și tulburările neurodegenerative și programul de donare a creierului și a corpului. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Structura Α-sinucleinei, modificarea posttranslațională și splicingul alternativ ca potențatori de agregare. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | Text integral | Google Scholar
Beyer, K., și Ariza, A. (2012). Modificarea posttranslațională a alfa-sinucleinei și splicingul alternativ ca factor declanșator al neurodegenerării. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., și Ariza, A. (2008). Expresia diferențială a izoformelor de alfa-sinucleină, parkin și sinfilină-1 în boala cu corpuri Lewy. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | Refef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., și Ariza, A. (2007). Identificarea și caracterizarea unei noi izoforme de alfa-sinucleină și rolul său în bolile cu corpuri Lewy. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Niveluri scăzute de ARNm al alfa-sinucleinei 126 în demența cu corpi Lewy și boala Alzheimer. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., și Filipowicz, W. (2010). Reglarea traducerii și stabilității ARNm prin microARN-uri. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: aliniere de secvențe multiple cu mare precizie și randament ridicat. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., și Kalyanaraman, B. (2010). Oxidanții induc splicingul alternativ al Α-sinucleinei: implicații pentru boala Parkinson. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., și Halliday, G. M. (2014). Biologia alfa-sinucleinei în bolile cu corpuri Lewy. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Varianta AR-V9 a receptorului de androgeni este coexprimată cu AR-V7 în metastazele cancerului de prostată și prezice rezistența la abirateron. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., și Lee, S. J. (2001). Α-sinucleina legată de membrană are o înclinație mare de agregare și capacitatea de a însămânța agregarea formei citosolice. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., și Chiba-Falek, O. (2015). Un haplotip bogat în citosină-timină (CT) în intronul 4 al SNCA conferă risc pentru patologia corpului Lewy în boala Alzheimer și afectează expresia SNCA. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosticul și managementul demenței cu corpi Lewy: al treilea raport al consorțiului DLB. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., și Perry, R. H. (1999). Raport al celui de-al doilea atelier internațional privind demența cu corpuri Lewy: diagnostic și tratament. Consorțiul privind demența cu corpuri Lewy. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., și Isacson, O. (2012). Nivelurile de expresie a transcripției alfa-sinucleinei de lungime completă și a celor trei variante de splicing alternativ ale acesteia în regiunile cerebrale ale bolii Parkinson și într-un model de șoarece transgenic de supraexprimare a alfa-sinucleinei. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scalarea descoperirii exacte a variantelor genetice precise la zeci de mii de eșantioane. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Utilizarea alternativă a transcripției Α-sinucleinei ca mecanism convergent în patologia bolii Parkinson. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Profilarea transcriptomică a neuronilor dopaminergici purificați derivați de pacienți identifică perturbații convergente și terapeutice pentru boala Parkinson. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., și Südhof, T. C. (2014). Cartografia splicingului alternativ al neurexinei cartografiată prin secvențierea ARNm cu citire lungă cu o singură moleculă. Proc. Natl. acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract |Ref Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., și Tassone, F. (2017). Expresia alterată a peisajului variantelor de splicing FMR1 la purtătorii de premutație. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). Legătura dintre gena SNCA și parkinsonism. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Poliadenilarea alternativă a ARNm: Regiunea 3′-untranslated contează în exprimarea genelor. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar
.