- Introduktion
- Materialer og metoder
- Studieprøver
- Genomisk DNA- og RNA-udtræk
- Library Preparation and Sequencing
- gDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
- DNA-indfangning ved hjælp af IDT Xgen® Lockdown®-prober og Single-Molecule Isoform-Sequencing (Iso-Seq)
- gDNA-analyse
- Short Variant Analysis and Phasing
- Clustering og bestemmelse af haplotyper for CT-Rich Region
- Isoformanalyse
- Isoform SNP Calling
- Resultater
- Targeted gDNA Capture identificerede kendte og nye variationer
- Targeted cDNA Capture Identified Novel Start and End Sites
- Fuld længde cDNA muliggør information om fasning på isoformniveau
- Diskussion
- Datatilgængelighed
- Author Contributions
- Funding
- Interessekonflikterklæring
- Akkreditering
- Supplementært materiale
Introduktion
Transkriptionelle og posttransskriptionelle programmer kontrollerer genekspressionsniveauer og/eller produktion af flere forskellige mRNA-isoformer, og ændringer i disse mekanismer resulterer i dysregulering af genekspression og differentielle ekspressionsprofiler. Aberrant transkriptionel og posttranskriptionel genregulering er hyppigt forekommende i væv i det menneskelige nervesystem og bidrager til fænotypiske forskelle inden for og mellem individer i sundhed og sygdom.
Dysregulering af alpha-synuclein-ekspression er blevet impliceret i patogenesen af synucleinopatier, især Parkinsons sygdom (PD) og demens med Lewy-kroppe og (DLB). Mens den rolle, som SNCA-overekspression spiller i synucleinopatier, hovedsagelig PD, er veletableret, har vi her fokuseret på bestemmelse af det komplette repertoire af SNCA-transkriptisoformer i forskellige synucleinopatier. Tidligere er flere forskellige SNCA-transkript isoformer blevet beskrevet for SNCA-genet, opstået fra alternativ splejsning, transkriptionelle startsteder (TSS’er) og udvælgelse af polyadenyleringssteder (McLean et al., 2012; Xu et al., 2014). Alternativ splejsning af de kodende exoner giver anledning til SNCA 140, SNCA 112, SNCA 126 og SNCA 98, hvilket resulterer i fire proteinisoformer (Beyer og Ariza, 2012). Alternative TSS’er i SNCA-genet resulterer i fire forskellige 5′UTR’er, og alternativ udvælgelse af forskellige polyadenyleringssteder bestemmer tre hovedlængder af 3′UTR’en, uden at det har nogen indvirkning på proteinproduktets sammensætning (Beyer og Ariza, 2012). Vores overordnede mål er at få ny indsigt i bidraget fra de forskellige SNCA mRNA-arter, kendte og nye, til patogenese og heterogenitet af synucleinopatier.
Til dato har de fleste undersøgelser brugt short read sekventeringsteknologier til at udspørge transkriptomkompleksiteten i menneskelige hjerner. Tilgængeligheden af tredje generation af long read-teknologier giver et hidtil uset og næsten fuldstændigt billede af isoformstrukturer. Imidlertid har eksisterende long read transkriptsekventering for humane sygdomsgener anvendt en ampliconbaseret tilgang (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Selv om denne tilgang har været vellykket med hensyn til at identificere kompleks alternativ splejsning i humane sygdomsgener, er den begrænset til PCR-primerdesignet og vil ikke afdække alternative start- og slutsteder. Målrettet berigelse, f.eks. ved brug af IDT-sonder, kan give en omfattende isoformvisning af gener af interesse til lave sekventeringsomkostninger. Endvidere muliggør meget nøjagtige læsninger af transkriptet i fuld længde isoform-specifik haplotyping.
Her præsenterer vi den første kendte undersøgelse ved hjælp af målrettet indfangning af gDNA og cDNA af SNCA-genregionen ved hjælp af PacBio SMRT-sekventering. SNCA-genregionen er ~114 kb lang og består af seks exoner med transkriptlængder på omkring 3 kb. Vi multiplexede 12 humane hjerneprøver fra PD, DLB og normale kontrolprøver og sekventerede gDNA- og cDNA-biblioteket på PacBio Sequel-systemet. Vi beskriver de bioinformatiske analyser, der blev anvendt til at identificere SNP’er, indels og korte tandemrepeats for gDNA-fangst og haplotyping på isoformniveau for cDNA-dataene. Vi viser, at målrettet indfangning er en omkostningseffektiv måde at studere genomisk variation og alternativ splejsning i et sygdomsrelateret neuralt gen i fællesskab.
Materialer og metoder
Studieprøver
Studiekohorten (N = 12) bestod af personer med tre obduktionsbekræftede neuropatologiske diagnoser: (1) PD (N = 4); (2) DLB (N = 4); og (3) klinisk og neuropatologisk normale personer (N = 4). Frontal cortex-hjernevæv blev opnået gennem Kathleen Price Bryan Brain Bank (KPBBB) på Duke University, Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) og Layton Aging and Alzheimer’s Disease Center på Oregon Health and Science University. Neuropatologiske fænotyper blev bestemt ved postmortemundersøgelse efter standard veletablerede metoder i overensstemmelse med McKeith og kollegers anbefalinger om metode og klinisk praksis (McKeith et al., 1999, 2005). Tætheden af LB-patologien (i et standardsæt af hjerneområder) fik scorer på mild, moderat, alvorlig og meget alvorlig. Undersøgelsesprøverne inden for hver diagnosegruppe, PD og DLB, blev omhyggeligt udvalgt på en sådan måde, at sværhedsgraden af de klinisk-patologiske fænotyper var ens inden for hver patologi. Alle hjerner udviste hjernestamme, limbiske og neokortikale Lewy bodies (LB’er), mens PD viste alvorlige til meget alvorlige McKeith-scoringer i sub-nigra og amygdala. Alle hjerner viser ingen AD i henhold til CERAD-kriterierne og Braak- og Braak-stadiet = II. De neurologisk sunde hjerneprøver blev udtaget fra postmortale væv fra klinisk normale personer, som i de fleste tilfælde blev undersøgt inden for et år efter dødsfaldet, og som viste sig ikke at have nogen kognitiv forstyrrelse eller parkinsonisme og neuropatologiske fund, der ikke var tilstrækkelige til at diagnosticere PD, Alzheimers sygdom (AD) eller andre neurodegenerative sygdomme. Alle prøver var hvide. Demografiske data og neuropatologi for disse forsøgspersoner er opsummeret i Supplerende tabel 1. Projektet blev godkendt af Duke Institution Review Board (IRB), som gav en etisk godkendelse. Metoderne blev udført i overensstemmelse med de relevante retningslinjer og bestemmelser.
Genomisk DNA- og RNA-udtræk
Genomisk DNA blev ekstraheret fra hjernevæv ved hjælp af standardprotokollen fra Qiagen (Qiagen, Valencia, CA). Samlet RNA blev ekstraheret fra hjerneprøver (100 mg) ved hjælp af TRIzol-reagens (Invitrogen, Carlsbad, CA) efterfulgt af rensning med et RNeasy-kit (Qiagen, Valencia, CA) i henhold til producentens protokol. gDNA- og RNA-koncentrationen blev bestemt spektrofotometrisk, og RNA-prøvernes kvalitet og mangel på væsentlig nedbrydning blev bekræftet ved målinger af RNA-integritetsnummeret (RIN, supplerende tabel 1) ved hjælp af en Agilent Bioanalyzer.
Library Preparation and Sequencing
gDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
Omkring 2 μg af hver gDNA-prøve blev skåret til 6 kb ved hjælp af Covaris g-TUBE og ligeret med stregkodede adaptere. En ækvimolær pulje af 12-plex stregkodet gDNA-bibliotek (2 μg i alt) blev indtastet i den sondebaserede indfangning med et specialudviklet SNCA-genpanel.
Et SMRTBell-bibliotek blev konstrueret ved hjælp af 626 ng af indfanget og re-amplificeret gDNA1.
DNA-indfangning ved hjælp af IDT Xgen® Lockdown®-prober og Single-Molecule Isoform-Sequencing (Iso-Seq)
Omkring 100-150 ng total RNA pr. reaktion blev omvendt transskriberet ved hjælp af Clontech SMARTer cDNA-syntesesættet og 12 prøvespecifikke stregkodede oligo dT (med PacBio 16mer stregkodesekvenser, se Supplerende metoder). Der blev behandlet tre omvendte transskriptioner (RT-reaktioner) parallelt for hver prøve. PCR-optimering blev anvendt til at bestemme det optimale antal amplifikationscyklusser for nedstrøms PCR-reaktioner i stor skala. En enkelt primer (primer IIA fra Clontech SMARTer-kittet 5′ AAG CAG CAG TGG TGG TAT CAA CGC CGC AGA GTA C 3′) blev anvendt til alle PCR-reaktioner efter RT. PCR-produkter i stor skala blev renset separat med 1X AMPure PB-beads, og bioanalysatoren blev anvendt til QC. En ækvimolær pulje af 12-plex stregkodet cDNA-bibliotek (1 μg i alt) blev indtastet i den sondebaserede indfangning med et specialudformet SNCA-genpanel.
Et SMRTBell-bibliotek blev konstrueret ved hjælp af 874 ng indfanget og re-amplificeret cDNA2. En SMRT Cell 1M (6 h film) blev sekventeret på PacBio Sequel-platformen ved hjælp af 2.0-kemi.
gDNA-analyse
Sekventering af de stregkodede gDNA-data blev kørt på tre SMRT Cells 1M ved hjælp af 2.0-kemi. Dataene blev demultiplexet ved at køre programmet Demultiplex Barcodes i PacBio SMRT Link v6.0.
Short Variant Analysis and Phasing
Circular Consensus Sequence (CCS)-reads blev genereret ved hjælp af SMRT Analysis 6.0 fra hvert demultiplexet datasæt og justeret til hg19-referencegenomet ved hjælp af minimap2. PCR-duplikater fra forstærkning efter fangst blev identificeret ved at kortlægge endepunkter og mærket ved hjælp af et brugerdefineret script. Korte varianter blev kaldt ved hjælp af GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Efter en første passage af filtrering ved hjælp af dækningsdybde og kvalitetsmetrikker blev varianter manuelt inspiceret i IGV3. Hvis varianter ikke var i fase med nærliggende SNP’er, blev de manuelt filtreret. De variantsteder, der passerede manuel kuratering, blev brugt sammen med de deduplikerede CCS-justeringer til read-backed phasing med WhatsHap (Martin et al., 2016).
Clustering og bestemmelse af haplotyper for CT-Rich Region
Subsekvenser, der er justeret til chr4: 90742331-90742559 (hg19), blev ekstraheret for hver prøve. Efter at have inspiceret størrelsesfordelingen af disse undersekvenser blev de grupperet efter størrelse og sekvenslighed ved hjælp af en kombination af python og MUSCLE (Edgar, 2004), og en konsensussekvens blev genereret uafhængigt for hver klynge.
Specifikke scripts og arbejdsgange er yderligere beskrevet i https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Isoformanalyse
Sekventering af de stregkodede cDNA-data blev foretaget på en SMRT Cell 1M på PacBio Sequel-systemet ved hjælp af 2.0-kemi. Bioinformatisk analyse blev udført ved hjælp af IsoSeq3-applikationen i PacBio SMRT Analysis v6.0.0.0 for at opnå isoform-sekvenser i fuld længde af høj kvalitet (se Supplerende metoder for flere oplysninger).
Isoform SNP Calling
Fuldlængde læsninger forbundet med de endelige 41 isoformer fra alle 12 prøver blev justeret til hg19-genomet for at skabe en pileup. Baser med QV på mindre end 13 blev udelukket. Derefter blev der på hver position med mindst 40 basers dækning anvendt en Fisher exact-test med Bonferroni-korrektion med en p-cutoff på 0,01. Kun substitutions-SNP’er, der ikke ligger tæt på homopolymerregioner (strækninger med 4 eller flere af det samme nukleotid), blev kaldt. Efter SNP-kaldning blev genotypen for hver prøve bestemt ved at tælle antallet af understøttende prøvespecifikke fuldlængde (FL) læsninger (FL-reads). Hvis en prøve havde 5+ FL-reads, der understøttede både reference- og alternativ base, var den heterozygot. Hvis en prøve havde 5+ FL-reads, der understøttede den ene allel, og færre end 5 FL-reads for den anden allel, var den homozygot. I modsat fald var den ikke entydig. Skripter er tilgængelige på: https://github.com/Magdoll/cDNA_Cupcake.
Resultater
Vi designede brugerdefinerede prober til SNCA-genet og udførte målrettet indfangning af både gDNA og cDNA på et multiplexet bibliotek bestående af 12 menneskelige hjerneprøver fra PD, DLB og normale kontroller (Figur 1, Supplerende tabel 1). GDNA- og cDNA-bibliotekerne blev sekventeret på PacBio Sequel-platformen. Bioinformatisk analyse blev udført ved hjælp af PacBio-software efterfulgt af brugerdefinerede analyser.
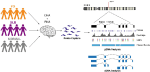
Figur 1. Skematisk præsentation af undersøgelsesdesignet. DNA- og RNA-materialer blev ekstraheret fra postmortale hjernevæv fra patienter med Parkinsons sygdom, demens med Lewy Body og kontrolgrupper. gDNA- og cDNA-biblioteker blev fremstillet ved hjælp af sondehybridisering og sekventeret på PacBio Sequel-systemet. Analyse blev udført ved hjælp af PacBio-software og andre eksisterende værktøjer.
Targeted gDNA Capture identificerede kendte og nye variationer
Efter generering af cirkulære konsensus-sekvenser (CCS) og fjernelse af PCR-duplikater (Supplerende metoder) opnåede vi 16- til 71-foldig gennemsnitlig unik dækning af SNCA-genregionen. CCS-læsningerne havde en gennemsnitlig indsætningslængde på 2,9 kb og en gennemsnitlig læsnøjagtighed på 98,9 %. Med undtagelse af et område på 5 kb, der bevidst blev afdækket af sonder på grund af tilstedeværelsen af LINE-elementer (hg19 chr4: 90697216-90702113) og et område på 2,1 kb med højt GC-indhold omkring exon 1, var der tilstrækkelig dækning til at genotype begge haplotyper for hver af de 212 prøver (Figur 2, Supplerende figur 1).

Figur 2. Målrettet gDNA-indfangning og fasning. Et eksempel, der viser en prøve fra hver tilstand. Øverste spor viser en af SNCA-isoformerne, efterfulgt af gDNA-dækningen for de tre prøver. Variantsporet viser hver SNP og er farvekodet for heterozygot (lilla), homozygot alternativ (orange) og homozygot reference (grå). Faserede blokke er vist med lyseblå. Det nederste spor viser placeringen af opsamlingssonder. Dropout-regionen i sondedesignet skyldes to LINE-elementer i midten af intron 4. For gDNA-dækning og fasningsoplysninger for alle 12 prøver, se Supplerende figurer.
Ved hjælp af GATK4 HC, kvalitetsbaseret filtrering og manuel kuratering identificerede vi 282 SNP’er og 35 indels, herunder 8 SNPS og 13 indels, der ikke findes i dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (Supplerende tabel 2). Der blev ikke identificeret nogen varianter i den kodende region for SNCA, selv om der blev identificeret otte varianter i utranslaterede regioner. Størstedelen af de identificerede varianter, herunder flere korte tandem gentagelser (STR), falder inden for intron 2, 3 og 4.
Vi har tidligere beskrevet et meget polymorft CT-rigt område i intron 4 af SNCA med fire observerede haplotyper (Lutz et al., 2015). Mens denne meget repetitive og strukturelt variable region viste sig at være vanskelig at genotype med GATK4 HC, var vi i stand til at konstruere konsensussekvenser for alle 12 prøver og observerede alle 4 af de tidligere opdagede haplotyper (Supplerende figur 2). Derudover identificerede vi en ny STR i intron 4, der består af en enhed på tre baser, der gentages 16 gange i referencen. Inden for de 12 prøver identificerede vi tre haplotyper med 9, 12 og 15 kopier af TTG gentagelsesenheden. GATK HC genotypede alle disse korrekt med undtagelse af en haplotype for PD-4, som havde ret lav dækning i denne region. Med de givne data for denne prøve kan genotypen imidlertid bestemmes ved visuel inspektion (tabel 1).

Tabel 1. En ny triplet tandem gentagelse i intron 4 (chr4: 90713442).
Vi brugte de korte varianter, der blev detekteret af GATK HC i forbindelse med det læsebaserede faseringsværktøj WhatsHap (Martin et al., 2016) til at fase CCS-læsningerne på tværs af locus, med en række succeser drevet hovedsagelig af den heterozygote varianttæthed over locus. Prøverne PD-1, PD-4, N-4, DLB-1 og DLB-4 havde lange strækninger med lav heterozygotitet med meget få, korte faseblokke, mens de andre prøver gav faseblokke, der varierede fra 7 til 18 gange den gennemsnitlige læselængde, op til 54 kb (Supplerende figur 3).
Targeted cDNA Capture Identified Novel Start and End Sites
Vi behandlede PacBio cDNA (Iso-Seq) dataene ved hjælp af PacBio SMRT Analysis software. Efter kortlægning af Iso-Seq-dataene til hg19 og fjernelse af artefakter (Supplerende tabel 3, Supplerende figur 4) opnåede vi et endeligt sæt af 41 SNCA-isoformer (figur 3). Alle endelige isoformer har alle de kanoniske splejsesteder (GT-AG eller GC-AG) og understøttes af i alt 20 eller flere læsninger i fuld længde. Størstedelen af isoformerne (28 ud af 41) har alle seks exoner og adskiller sig kun ved brug af alternative 5′-startsteder og 3′ UTR-længder. 3′ UTR-længderne varierede mellem 300 og 2,6 kb. Brugen af meget forskellige alternative 5′-startsteder i SNCA er kendt; hvad der er mindre kendt er den variable 3′ UTR-længde, som tidligere er blevet undersøgt ved hjælp af RNA-seq-data, der ikke opløste isoformstrukturer i fuld længde (Rhinn et al., 2012). Iso-Seq-dataene viser, at den variable 3′ UTR-længde synes at være parret med alle mulige kombinationer af 5′-startsteder uden nogen præferentiel kobling. Næsten ingen af variabiliteten i start- og slutsted ændrer den forudsagte åbne læseramme (Supplerende figur 5) og forudsiges at oversætte til den kanoniske 141 aminosyresekvens.

Figur 3. SNCA-isoformer, der er fanget ved hjælp af målrettet Iso-Seq, identificerer nye start- og slutsteder. Størstedelen af isoformkompleksiteten stammer fra kombinatorisk brug af alternative 3′ UTR-længder og exon 1, med nogle få sjældne alternative splejsesteder fundet i exon 1 (grøn), 2 (rød) og 4 (blå). Alle krydsninger har kanoniske splejsesteder. Vi identificerede fem isoformer, der sprang exon 5 over, og to isoformer, der sprang exon 3 over. Vi identificerede også nye start- (orange) og slutsteder (lilla) i intron 4. Kaldte SNP’er er markeret med lilla.
Vi validerede yderligere de nye (men kanoniske) forgreninger ved hjælp af offentligt tilgængelige kortlæste forgreningsdata. Intropolis (v1, https://github.com/nellore/intropolis) databasen kombinerer over 21.000 offentligt tilgængelige RNA-seq. På grund af den store mængde af junction-data, der kun understøttes af en enkelt short read, kræver vi i denne undersøgelse mindst 10 short read-støtte (kombineret fra alle >21.000 RNA-seq-datasæt) for at bekræfte vores Iso-Seq-nye junctions. Med undtagelse af de nye krydsninger for PB.1016.253 og PB.1016.296 (figur 3) understøttes alle andre nye krydsninger af Intropolis-datasættet. Det er interessant, at disse nye junctions har betydeligt mindre støtte fra korte læsninger end de Gencode-annoterede junctions. For eksempel understøttes de to nye junctions i PB.1016.139, der er introduceret af det nye exon, af henholdsvis 2.519 og 44 Intropolis-kortlæsninger, mens de fire andre kendte junctions understøttes af over 1 million kortlæsninger. Dette viser styrken af målrettet berigelse ved hjælp af sekventering af transkriptom i fuld længde til påvisning af sjældne, nye isoformer.
Vi observerede to isoformer med exon 3 skipping (SNCA126) og fem isoformer med exon 5 skipping (SNCA112). Igen stammer splejningsdiversiteten i disse to exon-skipping-grupper hovedsagelig fra den forskellige brug af alternative 5′-startsteder og variabel 3′ UTR-længde. ORF-prædiktion viser, at springning af exon 3 eller exon 5 forkorter ORF’en, men bibeholder læserammen. Tre isoformer har nye 3′-endesteder placeret i intron 4. ORF-prædiktion viser, at dette resulterer i et afkortet proteinprodukt.
Vi identificerede et tidligere ikke-annoteret 5′-startsted placeret i intron 4 (hg19 chr4: 90692548-90693045, figur 3). De tre isoformer, der er forbundet med denne nye start, består af det nye startsted, exon 5 og variable 3′ UTR-længder. Det er interessant, at mens offentligt downloadede short read-data fra GTEx og Sandor et al. (2017) og CAGE-topdata (FANTOM5) ikke understøttede dette nye startsted, indeholdt et nyligt offentligt NA12878 direkte RNA-datasæt4 kun ét SNCA-transkript, der bekræftede dette alternative startsted. Endvidere bekræftes den nye krydsning mellem exon 5 og det nye startsted af Intropolis short read junction-data. Det er interessant, at dette nye 5′-startsted forudsiges at introducere nye peptider, samtidig med at læsningsrammen i exon 5 opretholdes.
Vi identificerede også tre SNCA-transskriptioner med nye slutsteder (figur 3). To isoformer (PB.1016.383, PB.1016.384) anvendte en udvidet 3′ UTR i exon 4, mens den tredje isoform (PB.1016.381) anvendte et nyt 3′ exon i intron 4. De nye junctions mellem det nye sidste exon og det foregående exon understøttes af offentlige short read junction-data (Intropolis). De nye 3′ UTR’er resulterer i en trunkeret ORF-prædiktion.
Ved anvendelse af det normaliserede antal læsninger i fuld længde som en proxy for isoformens hyppighed finder vi, at en af de kanoniske SNCA-isoformer (PB.1016.131) er den mest hyppige, med en hyppighed på 50-60 % på tværs af alle prøveemner (Supplerende tabel 4). Vi grupperede yderligere de 41 isoformer efter deres splejsningmønstre (tabel 2). Isoformer, der har alle seks exoner, tegner sig for 95-97 % af hyppigheden. Tidligere undersøgelser har vist en markant stigning i ekspressionen af isoformer, der mangler exon 3 (SNCA126) i den frontale cortex af DLB-prøver sammenlignet med normale (Beyer et al., 2008); vores aggregerede isoformtællinger viser, at tre af DLB-prøverne har et let forhøjet tællingsniveau sammenlignet med de normale prøver samt SNCA112 (exon 5 skipping) varianter for PD og DLB i forhold til normale prøver.

Tabel 2. SNCA-isoforms overflod for hver prøve, aggregeret efter splejsningmønstre.
Fuld længde cDNA muliggør information om fasning på isoformniveau
Vi kaldte SNP’er ved hjælp af cDNA ved at stable alle fuldlængde læsninger fra de 12 prøver for at kalde varianter (se afsnittet “Metoder”). I alt fire SNP’er blev kaldt, og alle var tidligere annoteret i dbSNP (tabel 3, figur 3). De fire SNP’er er alle placeret i ikke-CDS-regioner, en i 3′ UTR (exon 6), en i intron 4 og to i 5′ UTR (exon 1). SNP’en i 3′ UTR (chr4: 90646886) er kun dækket af isoformer med en 3′ UTR, der er mindst ~1 kb lang, og det er derfor ikke alle kanoniske isoformer, der dækker denne SNP. SNP’en i intron 4 (chr4: 90743331) er kun dækket af de nye alternative 3′-ende isoformer (PB.1016.383, PB.1016.384) og er ikke forbundet med nogen af de andre SNP’er. De to 5′ UTR SNP’er (chr4: 90757312 og chr4: 90758389) er dækket af to gensidigt udelukkende exon 1-brug og er derfor heller ikke forbundet.

Tabel 3. cDNA SNP-oplysninger.
Vores nuværende fremgangsmåde er begrænset til kun at kalde substitutionsvarianter i transskriberede regioner med tilstrækkelig dækning. Sammenligning af listen over vores SNP’er med hg19 dbSNP-annotationen viser, at de fleste af de SNP’er eller varianter, der blev overset, enten var mindre end 1 % hyppighed i befolkningen, ikke var enkelt nukleotid-substitutioner eller støder op til regioner med lav kompleksitet. For eksempel er rs77964369 (chr4: 90646532) rapporteret til at have en 50/50-frekvens af T/A; dette T støder imidlertid op til en strækning på 11 genomiske As nedstrøms. Manuel inspektion af Iso-Seq read pileup, som har ~ 1,300 læsninger på dette sted, tyder ikke på tegn på variation i det mindste blandt vores 12 prøver.
Ved hjælp af de prøvespecifikke læsninger kalder vi genotypen for hver prøve på hver SNP-placering (tabel 3). Udover PD-2, der har for få læsninger og er ukonklusiv for alle fire SNP’er, var vi i stand til at kalde genotypen for de fleste andre prøver. DLB-3 var især den eneste prøve, der er heterozygot på alle SNP-lokationer. Ellers observerede vi ikke noget tilstandsspecifikt mønster for at foretrække den ene genotype frem for den anden.
Diskussion
Vi beskriver den første undersøgelse, der anvender målrettet berigelse af SNCA-genet på multiplexede gDNA- og cDNA-biblioteker til undersøgelse af neurologiske sygdomme ved hjælp af long read-sekventering. De lange læselængder i PacBio Sequel-systemet gjorde det lettere at sekventere SNCA-genets fulde repertoire af transkript isoformer i fuld længde. Vi afslørede diversiteten i brugen af alternative 5′-startsteder og variable 3′ UTR-længder og observerede kendte exon-skipping-hændelser som f.eks. exon 3-deletion (SNCA126) og exon 5-deletion (SNCA112). Desuden blev der identificeret nye alternative start- og slutsteder inden for det store intron 4, som forudsiges at blive oversat til nye proteiner. Det er sandsynligt, at den store dybde af sekventeringsdækning ved målrettet opsamling i kombination med evnen til at sekventere komplette transskriptioner gjorde det muligt for os at opdage disse tidligere ubeskrevne isoformer.
Den biologiske og patologiske betydning af de forskellige SNCA-proteinisoformer er endnu ikke fuldt ud afdækket. Specifikke SNCA posttranslationsmodifikation og splejsning af isoformer er imidlertid blevet forbundet med intracellulære aggregationsproportioner (Kalivendi et al., 2010) og udtrykkes forskelligt i menneskelige synucleinopatier (Beyer et al., 2008; Beyer og Ariza, 2012). Undersøgelser af SNCA posttranslationsmodifikation viste, at Lewy bodies, det patologiske kendetegn for synucleinopatier, indeholder rigeligt fosforyleret, nitreret og monoubiquitineret SNCA (Kim et al., 2014). Virkningerne af post-transkriptionel modifikation på SNCA-aggregation er også blevet undersøgt. Alternativ splejsning blev foreslået for at påvirke SNCA-aggregation. En deletion af enten exon 3 eller 5 forudsiger funktionelle konsekvenser: mens exon 3-deletion (SNCA126) fører til afbrydelse af det N-terminale protein-membraninteraktionsdomæne, hvilket kan føre til mindre aggregering, og exon 5-deletion (SNCA112) kan resultere i øget aggregering på grund af en betydelig forkortelse af den ustrukturerede C-terminus (Lee et al., 2001; Beyer, 2006). I den frontale cortex af DLB er SNCA112 markant forøget i forhold til kontrollerne (Beyer et al., 2008), mens SNCA126-niveauet er nedsat i den præfrontale cortex hos DLB-patienter (Beyer et al., 2006). I modsætning hertil viste SNCA126-ekspressionen øget i den frontale cortex i PD-hjerner og ingen signifikante forskelle i MSA (Beyer et al., 2008). SNCA98 er en hjernespecifik splejsningsvariant, der mangler både exon 3 og 5 og udviser forskellige ekspressionsniveauer i forskellige områder af foster- og voksenhjernen. Overekspression af SNCA98 er blevet rapporteret i DLB, PD (Beyer et al., 2007) og MSA (Beyer et al., 2008) frontale cortexer sammenlignet med kontroller. Desuden blev den posttranskriptionelle proces, der resulterer i alternativ 3′UTR-brug, rapporteret at have virkninger på mRNA-stabilitet og lokalisering (Fabian et al., 2010; Rhinn et al., 2012; Yeh og Yong, 2016). Yderligere undersøgelser vedrørende aggregeringspropendens af de forskellige kendte SNCA-proteinisoformer og sammensætningen af Lewy bodies er berettiget. Desuden lagde vores undersøgelse grundlaget for mRNA-kvantificeringsanalyser af de tidligere kendte og nye transskriptioner i en større stikprøvestørrelse bestående af emner med en række klinisk-patologiske stadier ved hjælp af flere hjerneområder fra hvert emne. Disse analyser af det hjerneregionsspecifikke transkriptomiske landskab af SNCA i forbindelse med neuropatologisk sværhedsgrad vil være informative med hensyn til den rolle, som specifikke SNCA-transkriptisoformer spiller i progressionen af de neuropatologiske stadier og sværhedsgraden af Lewy bodies og Lewy-neuritternes tæthed.
I denne artikel fokuserede vi på at skabe en sekventerings- og analysestandard for analyse af målrettede gDNA- og cDNA-data genereret fra de samme emner. Dette er en kraftfuld tilgang, der potentielt gør det muligt at faser gDNA-sekvenserne på tværs af hele regionen af et bestemt gen baseret på heterozygositet i sekvensen af isoformerne af det fulde transkript i fuld længde. De PacBio målrettede gDNA-data i denne undersøgelse producerede fasede blokke, der dækkede 81 % af den 114 kb store region centreret om SNCA, med den længste fasede blok på over 54 kb. Da gDNA-fasering er begrænset af læselængde og heterozygositet, vil stigende læselængder sandsynligvis generere større faseblokke.
gDNA-variantanalyse bekræftede kendte og identificerede nye korte tandem gentagelser (STR’er) i de introniske regioner. For eksempel opdagede vi tidligere ved hjælp af fasesekvent sekventering ved kloning og Sanger-sekventering fire forskellige haplotyper inden for en intronisk CT-rige region, der bestod af en klynge af variable gentagne sekvenser (Lutz et al., 2015). Vi viste, at en specifik haplotype, kaldet haplotype 3, gav risiko for at udvikle Lewy body-patologi hos Alzheimers patienter. Her validerede vi sekvensen af denne meget polymorfe lavkomplekse region med lav kompleksitet og dens fire definerede haplotyper. Selv om vores stikprøve var lille, var “haplotype 3” udelukkende til stede hos sygdomspatienter (en PD-patient, to DLB-patienter), hvilket er i overensstemmelse med vores tidligere resultater. Pilotresultaterne og vores tidligere publikation giver forudsætningen for at gentage foreningsanalyserne af synucleinopatier med præcist definerede, dvs. ved lange læsninger, STR’er og strukturelle haplotyper ved hjælp af en større stikprøvestørrelse.
Vores artikel demonstrerede PacBio Sequel-systemets evne til at opdage nye fuldlængde-transskriptioner og karakterisere det komplette fuldlængde-transskriptrepertoire for et gen, der er impliceret i en sygdom. Desuden viste vi også, at gDNA med lange læsninger definerer korte strukturelle varianter og haplotyper, herunder STR’er, mere præcist og derved kan lette opdagelsen og valideringen af andre sygdomsrelaterede varianter end SNP’er. Samlet set er denne nye viden meget værdifuld og anvendelig til at fremme vores forståelse af de genetiske ætiologier, som kan involvere forstyrrelser i transkriptlandskabet, der ligger til grund for komplekse menneskelige sygdomme, herunder aldersrelaterede neurodegenerative sygdomme som f.eks. synucleinopatier.
Datatilgængelighed
De tre SMRT-celler af gDNA-råddata er tilgængelige på Zenodo.org med doi: 10.5281/zenodo.1560688. Den ene SMRT-celle af cDNA-råddata er tilgængelig på Zenodo.org med doi: 10.5281/zenodo.1581809. De behandlede gDNA- og cDNA-resultater, herunder gDNA-varianter og cDNA-isoformer, er tilgængelige på Zenodo.org med doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F bidrog til udformning og design af undersøgelsen. ET og WR organiserede sekvensdatabaser, udførte sekventeringsanalyserne og udarbejdede alle figurer og tabeller. O-CG og JB håndterede hjernevæv og præparater af nukleinprøver. TH genererede sekventeringsdatasættene. SK designede og skaffede reagenser. OC-F, ET og WR skrev det første udkast til manuskriptet. OC-F opnåede finansiering. Alle forfattere bidrog til forberedelse af manuskriptet, læste og godkendte den indsendte version.
Funding
Dette arbejde blev delvist finansieret af National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Interessekonflikterklæring
ET, WR, TH og SK er eller var ansatte hos Pacific Biosciences på tidspunktet for undersøgelsen.
De resterende forfattere erklærer, at forskningen blev udført uden kommercielle eller finansielle relationer, der kunne opfattes som en potentiel interessekonflikt.
Akkreditering
Dette manuskript er blevet udgivet som et pre-print på BioRxiv (Tseng et al, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Supplementært materiale
Det supplerende materiale til denne artikel kan findes online på: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona undersøgelse af aldring og neurodegenerative sygdomme og hjerne- og kropsdonationsprogram. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-synuclein struktur, posttranslationel modifikation og alternativ splicing som aggregationsforstærkere. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., og Ariza, A. (2012). Alpha-synuclein posttranslationel modifikation og alternativ splejsning som en udløsende faktor for neurodegeneration. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-833030-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., og Ariza, A. (2008). Differentiel ekspression af alfa-synuclein, parkin og synphilin-1 isoformer i Lewy body disease. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., og Ariza, A. (2007). Identifikation og karakterisering af en ny alfa-synuclein isoform og dens rolle i Lewy body sygdomme. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Lave alpha-synuclein 126 mRNA-niveauer i demens med Lewy-kroppe og Alzheimers sygdom. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., og Filipowicz, W. (2010). Regulering af mRNA-oversættelse og stabilitet af mikroRNA’er. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multipel sekvenstilpasning med høj nøjagtighed og høj gennemstrømning. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Oxidanter inducerer alternativ splejsning af Α-synuclein: implikationer for Parkinsons sygdom. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., og Halliday, G. M. (2014). Alpha-synuclein biologi i Lewy body sygdomme. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgenreceptorvariant AR-V9 er samudtrykt med AR-V7 i metastaser af prostatakræft og forudsiger abirateronresistens. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., og Lee, S. J. (2001). Membranbundet Α-synuclein har en høj aggregationstilbøjelighed og evnen til at sætte aggregering af den cytosoliske form i gang. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., og Chiba-Falek, O. (2015). En cytosin-thymin (CT)-rig haplotype i intron 4 af SNCA giver risiko for Lewy body-patologi i Alzheimers sygdom og påvirker SNCA-ekspression. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/08505050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosticering og behandling af demens med Lewy bodies: tredje rapport fra DLB-konsortiet. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., og Perry, R. H. (1999). Rapport fra den anden internationale workshop om demens med Lewy body: diagnose og behandling. Consortium on dementia with Lewy bodies. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., og Isacson, O. (2012). Transkriptudtryksniveauer af fuld længde alpha-synuclein og dets tre alternativt spliced varianter i Parkinsons hjerneområder og i en transgen musemodel af alpha-synuclein overekspression. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to ten of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternativ Α-synuclein transkriptbrug som en konvergerende mekanisme i Parkinsons sygdomspatologi. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transkriptomisk profilering af rensede patientafledte dopaminneuroner identificerer konvergerende forstyrrelser og terapeutiske midler til Parkinsons sygdom. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., og Südhof, T. C. (2014). Kartografi af neurexin alternativ splejsning kortlagt ved enkeltmolekyle long-read mRNA-sekventering med enkeltmolekyle. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). Landskabet af SNCA-transskriptioner på tværs af synucleinopatier: ny indsigt fra long reads sekventeringsanalyse. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., og Tassone, F. (2017). Ændret udtryk af FMR1-splejningsvarianternes landskab i præmutationsbærere. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., og Yu, J.-T. (2014). Forbindelsen mellem SNCA-genet og parkinsonisme. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., og Yong, J. (2016). Alternativ polyadenylering af mRNA’er: 3′-untranslated region har betydning for genekspression. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar