- Introduction
- Materiaalit ja menetelmät
- Tutkimusnäytteet
- Genomisen DNA:n ja RNA:n uutokset
- Kirjaston valmistelu ja sekvensointi
- gDNA:n talteenotto IDT Xgen® Lockdown® -koettimien avulla ja yhden molekyylin sekvensointi
- cDNA:n kaappaus IDT Xgen® Lockdown® -koettimien avulla ja yhden molekyylin isomuodon sekvensointi (Iso-Seq)
- gDNA-analyysi
- Lyhyt muunnosanalyysi ja vaiheistus
- Clustering and Determining Haplotypes for CT-Rich Region
- Isoformianalyysi
- Isomuodon SNP-kutsu
- Tulokset
- Targeted gDNA Capture Identified Known and Novel Variations
- Kohdennettu cDNA-kaappaus identifioi uusia alku- ja loppusijoituspaikkoja
- Full-Length cDNA Enables Isoform-Level Phasing Information
- Keskustelu
- Datan saatavuus
- Author Contributions
- Rahoitus
- Eriintyneisyysristiriitoja koskeva lausunto
- Kiitokset
- Lisäaineisto
Introduction
Transkriptionaaliset ja posttranskriptionaaliset ohjelmat kontrolloivat geenien ilmentymistasoja ja/tai useiden erillisten mRNA-isoformien tuotantoa, ja muutokset näissä mekanismeissa johtavat geenien ilmentymisen häiriöihin ja eriytyneisiin ilmentymisprofiileihin. Transkriptionaalisen ja posttranskriptionaalisen geenisäätelyn poikkeavuuksia esiintyy runsaasti ihmisen hermostokudoksissa, ja ne vaikuttavat osaltaan fenotyyppisiin eroihin yksilöiden sisällä ja yksilöiden välillä sekä terveydessä että sairauksissa.
Alfa-synukleiinin ilmentymisen epäsäännöllisyys on yhdistetty synukleiinisairauksien patogeneesiin, erityisesti Parkinsonin taudin ja Lewyn kappaleisiin perustuvan dementian (Dementia with Lewy bodies and (DLB)). SNCA:n yliekspression merkitys synukleiinisairauksissa, lähinnä Parkinsonin taudissa, on todettu hyvin, mutta tässä tutkimuksessa keskityttiin SNCA:n transkriptioiden isomuotojen koko repertuaarin määrittämiseen eri synukleiinisairauksissa. Aiemmin SNCA-geenille on kuvattu useita erilaisia SNCA-transkriptin isoformeja, jotka ovat syntyneet vaihtoehtoisen pilkkomisen, transkription aloituskohtien (TSS) ja polyadenylaatiokohtien valinnan seurauksena (McLean ym., 2012; Xu ym., 2014). Koodaavien eksonien vaihtoehtoinen splikointi synnyttää SNCA 140:n, SNCA 112:n, SNCA 126:n ja SNCA 98:n, jolloin syntyy neljä proteiinin isoformia (Beyer ja Ariza, 2012). SNCA-geenin vaihtoehtoiset TSS:t johtavat neljään erilaiseen 5′UTR:ään, ja eri polyadenylaatiokohtien vaihtoehtoinen valinta määrittää 3′UTR:n kolme pääpituutta, joilla ei ole vaikutusta proteiinituotteen koostumukseen (Beyer ja Ariza, 2012). Yleistavoitteemme on saada uutta tietoa siitä, miten erilaiset SNCA:n mRNA-lajit, tunnetut ja uudet, vaikuttavat synukleinopatioiden patogeneesiin ja heterogeenisuuteen.
Tähän mennessä useimmissa tutkimuksissa on käytetty lyhyen lukusekvensoinnin tekniikoita ihmisaivojen transkriptomin monimutkaisuuden tutkimiseen. Kolmannen sukupolven pitkien lukutekniikoiden saatavuus tarjoaa ennennäkemättömän ja lähes täydellisen kuvan isoformien rakenteista. Nykyisissä ihmisen sairausgeenejä koskevissa pitkien lukujen transkriptiosekvensoinnissa on kuitenkin käytetty amplikonipohjaista lähestymistapaa (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Vaikka tämä lähestymistapa on onnistunut tunnistamaan monimutkaista vaihtoehtoista splikointia ihmisen sairausgeeneissä, se rajoittuu PCR-alukkeiden suunnitteluun eikä paljasta vaihtoehtoisia alku- ja loppukohtia. Kohdennetulla rikastamisella, esimerkiksi käyttämällä IDT-koettimia, voidaan tuottaa kattava isoforminäkymä kiinnostavista geeneistä alhaisilla sekvensointikustannuksilla. Lisäksi erittäin tarkat täyspitkän transkriptin lukemat mahdollistavat isoformispesifisen haplotyypityksen.
Tässä esittelemme ensimmäisen tunnetun tutkimuksen, jossa käytettiin SNCA-geenin alueen gDNA:n ja cDNA:n kohdennettua sieppausta PacBio SMRT -sekvensointia käyttäen. SNCA-geenin alue on ~114 kb pitkä, ja se koostuu kuudesta eksonista, joiden transkriptien pituudet ovat noin 3 kb. Multipleksoimme 12 ihmisen aivonäytettä PD-, DLB- ja normaaleista kontrollinäytteistä ja sekvensoimme gDNA- ja cDNA-kirjaston PacBio Sequel -järjestelmällä. Kuvaamme bioinformatiikan analyysit, joita käytettiin SNP:iden, indeleiden ja lyhyiden tandemtoistojen tunnistamiseen gDNA-kaappauksen osalta ja isoformitason haplotyypityksen cDNA-tietojen osalta. Osoitamme, että kohdennettu kaappaus on kustannustehokas tapa tutkia yhdessä genomista variaatiota ja vaihtoehtoista splikointia sairauteen liittyvässä hermostogeenissä.
Materiaalit ja menetelmät
Tutkimusnäytteet
Tutkimuskohortti (N = 12) koostui henkilöistä, joilla oli kolme ruumiinavauksessa vahvistettua neuropatologista diagnoosia: (1) Parkinson (N = 4); (2) DLB (N = 4); ja (3) kliinisesti ja neuropatologisesti normaalit koehenkilöt (N = 4). Etuaivokuoren aivokudokset saatiin Duken yliopiston Kathleen Price Bryan Brain Bankin (KPBBB), Banner Sun Health Research Institute Brain and Body Donation Programin (Beach et al., 2015) ja Layton Aging and Alzheimer’s Disease Centerin kautta Oregon Health and Science Universityssä. Neuropatologiset fenotyypit määritettiin post mortem -tutkimuksessa vakiintuneilla vakiomenetelmillä noudattaen McKeithin ja kollegoiden menetelmää ja kliinisiä käytäntösuosituksia (McKeith ym., 1999, 2005). LB-patologian tiheys (vakioiduilla aivoalueilla) sai pisteitä lievästä, kohtalaisesta, vakavasta ja erittäin vakavasta. Tutkimusnäytteet kussakin diagnoosiryhmässä, PD ja DLB, valittiin huolellisesti siten, että kliinispatologisten fenotyyppien vakavuus oli samanlainen kussakin patologiassa. Kaikissa aivoissa esiintyi aivorungon, limbisen ja neokortikaalisen Lewyn kappaleet (LB), kun taas Parkinsonin taudin (PD) subnigran ja amygdalan McKeith-pisteet vaihtelivat vakavista erittäin vakaviin. Kaikissa aivoissa ei CERAD-kriteerien ja Braakin ja Braakin vaiheen = II mukaan ollut Alzheimerin tautia. Neurologisesti terveet aivonäytteet saatiin post mortem -kudoksista kliinisesti normaaleilta koehenkilöiltä, jotka tutkittiin useimmissa tapauksissa vuoden kuluessa kuolemasta ja joilla ei todettu olevan kognitiivisia häiriöitä tai parkinsonismia eikä neuropatologisia löydöksiä, jotka eivät riittäisi Parkinsonin taudin, Alzheimerin taudin tai muiden hermoston rappeutumishäiriöiden diagnosoimiseksi. Kaikki näytteet olivat valkoihoisia. Näiden koehenkilöiden demografiset ja neuropatologiset tiedot on esitetty tiivistetysti lisätaulukossa 1. Hankkeen hyväksyi Duke Institution Review Board (IRB), joka antoi eettisen hyväksynnän. Menetelmät toteutettiin asiaankuuluvien ohjeiden ja määräysten mukaisesti.
Genomisen DNA:n ja RNA:n uutokset
Genominen DNA uutettiin aivokudoksista Qiagenin standardiprotokollalla (Qiagen, Valencia, CA). Kokonais-RNA uutettiin aivonäytteistä (100 mg) TRIzol-reagenssilla (Invitrogen, Carlsbad, CA), minkä jälkeen se puhdistettiin RNeasy-kitillä (Qiagen, Valencia, CA) valmistajan protokollan mukaisesti. gDNA:n ja RNA:n pitoisuudet määritettiin spektrofotometrisesti, ja RNA-näytteiden laatu ja merkittävän hajoamisen puuttuminen varmistettiin mittaamalla RNA:n eheyslukua (RIN, lisätaulukko 1) Agilent Bioanalysaattoria käyttäen.
Kirjaston valmistelu ja sekvensointi
gDNA:n talteenotto IDT Xgen® Lockdown® -koettimien avulla ja yhden molekyylin sekvensointi
Kustakin gDNA-näytteestä leikattiin noin 2 μg 6 kb:n pituiseksi Covaris g-TUBE:lla ja ligatoitiin viivakoodatuilla adaptereilla. Ekvimolaarinen pooli 12-pleksin viivakoodattua gDNA-kirjastoa (yhteensä 2 μg) syötettiin koettimiin perustuvaan kaappaukseen räätälöidyllä SNCA-geenipaneelilla.
SMRTBell-kirjasto rakennettiin käyttäen 626 ng kaapattua ja uudelleen monistettua gDNA:ta1.
cDNA:n kaappaus IDT Xgen® Lockdown® -koettimien avulla ja yhden molekyylin isomuodon sekvensointi (Iso-Seq)
Noin 100-150 ng kokonais-RNA:ta reaktiota kohti käänteistranskriboitiin Clontech SMARTer cDNA-synteesikitin ja 12 näytekohtaisen viivakoodatun oligo dT:n (PacBio 16mer-viivakoodisekvenssejä sisältävän oligo dT:n) kanssa (ks. lisämenetelmät). Kullekin näytteelle käsiteltiin rinnakkain kolme käänteistä transkriptioreaktiota (RT). PCR-optimointia käytettiin optimaalisen monistussyklin lukumäärän määrittämiseksi myöhemmille suurille PCR-reaktioille. Yhtä aluketta (Clontech SMARTer kitin aluketta IIA 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) käytettiin kaikissa RT:n jälkeisissä PCR-reaktioissa. Suuren mittakaavan PCR-tuotteet puhdistettiin erikseen 1X AMPure PB -helmillä, ja laadunvalvontaan käytettiin bioanalysaattoria. Ekvimolaarinen pooli 12-pleksin viivakoodattua cDNA-kirjastoa (yhteensä 1 μg) syötettiin koettimiin perustuvaan kaappaukseen räätälöidyllä SNCA-geenipaneelilla.
SMRTBell-kirjasto rakennettiin käyttämällä 874 ng kaapattua ja uudelleen monistettua cDNA2:ta. Yksi SMRT Cell 1M (6 h elokuva) sekvensoitiin PacBio Sequel -alustalla käyttäen 2.0-kemiaa.
gDNA-analyysi
Sekvensointi viivakoodatusta gDNA-datasta suoritettiin kolmella SMRT Cell 1M:llä käyttäen 2.0-kemiaa. Tiedot demultipleksattiin ajamalla Demultiplex Barcodes -sovellus PacBio SMRT Link v6.0:ssa.
Lyhyt muunnosanalyysi ja vaiheistus
Kustakin demultipleksatusta datasarjasta luotiin SMRT Analysis 6.0:n avulla CCS-lukemat (CCS = Circular Consensus Sequence), jotka linjattiin hg19-vertailugenomiin minimap2:n avulla. PCR-duplikaatit, jotka olivat peräisin sieppauksen jälkeisestä monistuksesta, tunnistettiin kartoittamalla päätepisteet ja merkittiin mukautetun skriptin avulla. Lyhyet variantit kutsuttiin käyttäen GATK4 HaplotypeCalleria (GATK4 HC) (Poplin et al., 2018). Ensimmäisen suodatuskierroksen jälkeen, jossa käytettiin kattavuussyvyyttä ja laatumittareita, variantit tarkastettiin manuaalisesti IGV3:ssa. Jos variantit eivät vaiheistuneet läheisten SNP:iden kanssa, ne suodatettiin manuaalisesti. Manuaalisen kuratoinnin läpäisseet varianttipaikat käytettiin yhdessä deduplikoitujen CCS-kohdistusten kanssa WhatsHap-ohjelmalla (Martin et al., 2016) tapahtuvaan lukupohjaiseen vaiheistukseen.
Clustering and Determining Haplotypes for CT-Rich Region
Kustakin näytteestä poimittiin alisekvenssejä, jotka oli kohdistettu kohtaan chr4: 90742331-90742559 (hg19). Kun näiden osasekvenssien kokojakauma oli tarkastettu, ne klusteroitiin koon ja sekvenssin samankaltaisuuden perusteella käyttämällä pythonin ja MUSCLE:n (Edgar, 2004) yhdistelmää, ja konsensussekvenssi luotiin itsenäisesti kullekin klusterille.
Muokattuja skriptejä ja työnkulkuja kuvataan tarkemmin kohdassa https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Isoformianalyysi
Sekvensointi viivakoodatusta cDNA-datasta tehtiin yhdellä SMRT Cell 1M:llä PacBio Sequel -järjestelmällä käyttäen 2.0-kemiaa. Bioinformatiikan analyysi tehtiin käyttäen PacBio SMRT Analysis v6.0.0:n IsoSeq3-sovellusta PacBio SMRT Analysis v6.0.0:n laadukkaan, täyspitkän isomuodon sekvenssien saamiseksi (ks. lisätiedot Supplementary Methods).
Isomuodon SNP-kutsu
Täysin pitkät luetut lukemat, jotka liittyivät lopulliseen 41 isomuotoon kaikilta kahdeltatoista näytteeltä, linjattiin yhteen hg19-genomin kanssa pileupin luomista varten. Basit, joiden QV oli alle 13, jätettiin pois. Sitten jokaisessa kohdassa, jossa oli vähintään 40 emäksen kattavuus, sovellettiin Fisherin tarkkaa testiä Bonferroni-korjauksen kanssa p-rajan ollessa 0,01. Ainoastaan substituutio-SNP:t, jotka eivät ole lähellä homopolymeerialueita (vähintään neljän saman nukleotidin pituisia jaksoja), kutsuttiin. SNP:n kutsumisen jälkeen kunkin näytteen genotyyppi määritettiin laskemalla yhteen näytekohtaisten kokopituuslukujen (FL) lukumäärä. Jos näytteessä oli yli 5 FL-lukua, jotka tukivat sekä viite- että vaihtoehtoista emästä, se oli heterotsygoottinen. Jos näytteessä oli yli 5 FL-lukua, jotka tukivat yhtä alleelia, ja alle 5 FL-lukua, jotka tukivat toista alleelia, näyte oli homotsygoottinen. Muussa tapauksessa se oli epäselvä. Skriptit ovat saatavilla osoitteessa: https://github.com/Magdoll/cDNA_Cupcake.
Tulokset
Suunnittelimme SNCA-geenille räätälöityjä koettimia ja suoritimme sekä gDNA:n että cDNA:n kohdennetun kaappauksen multipleksoidulle kirjastolle, joka koostui 12:sta Parkinsonin taudin (PD), DLB:n (DLB:n) ja normaalin kontrollin ihmisaivojen näytteestä (kuva 1, täydentävä taulukko 1). gDNA- ja cDNA-kirjastot sekvensoitiin PacBio Sequel -alustalla. Bioinformatiikan analyysi tehtiin PacBio-ohjelmistolla, jota seurasi mukautettu analyysi.
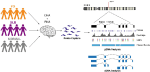
KUVIO 1. Bioinformatiikan analyysi. Kaavamainen esitys tutkimusasetelmasta. DNA- ja RNA-materiaalit uutettiin Parkinsonin tautia, Dementia with Lewy Body -tautia sairastavien potilaiden ja kontrolliryhmien post mortem -aivokudoksista. gDNA- ja cDNA-kirjastot valmistettiin koettimien hybridisaation avulla ja sekvensoitiin PacBio Sequel -järjestelmällä. Analyysi suoritettiin käyttäen PacBio-ohjelmistoa ja muita olemassa olevia työkaluja.
Targeted gDNA Capture Identified Known and Novel Variations
Sen jälkeen, kun olimme luoneet ympyränmuotoisia konsensussekvenssejä (Circle Consensus Sequences, CCS) ja poistaneet PCR-kaksoiskappaleet (Supplemental Methods), saimme SNCA-geenin alueelle 16-71-kertaisen keskimääräisen ainutlaatuisen peiton. CCS-lukujen keskimääräinen insertin pituus oli 2,9 kb ja keskimääräinen lukutarkkuus 98,9 %. Lukuun ottamatta 5 kb:n aluetta, joka tarkoituksellisesti jätettiin koettimilla peittämättä LINE-elementtien esiintymisen vuoksi (hg19 chr4: 90697216-90702113), ja 2,1 kb:n aluetta, jolla on korkea GC-pitoisuus eksoni 1:n ympärillä, kattavuus oli riittävä molempien haplotyyppien genotyypittämiseen jokaisessa 212 näytteessä (kuva 2, Täydentävä kuva 1).

Kuva 2. Kohdennettu gDNA:n talteenotto ja vaiheistus. Esimerkki, jossa näkyy yksi näyte kustakin tilasta. Ylimmällä raidalla näkyy yksi SNCA-isoformi, jonka jälkeen gDNA:n peitto kolmesta näytteestä. Varianttiraidassa näkyy kukin SNP, ja ne on värikoodattu heterotsygoottiseksi (violetti), homotsygoottiseksi vaihtoehtoiseksi (oranssi) ja homotsygoottiseksi referenssiksi (harmaa). Vaiheistetut lohkot on esitetty vaaleansinisellä. Alin rata osoittaa kaappauskoettimien sijainnit. Koettimien suunnittelussa esiintyvä pudotusalue johtuu kahdesta LINE-elementistä intronin 4 keskellä. Kaikkien 12 näytteen gDNA-peittävyys- ja vaiheistustiedot löytyvät täydentävistä kuvista.
Käyttämällä GATK4 HC:tä, laatupohjaista suodatusta ja manuaalista kuratointia tunnistimme 282 SNP:tä ja 35 indeliä, mukaan lukien 8 SNP:tä ja 13 indeliä, joita ei löydy dbSNP:stä (dbSNP Build ID: human_9606_b150_GRCh37p13) (täydentävä taulukko 2). SNCA:n koodaavalta alueelta ei tunnistettu yhtään varianttia, vaikka kahdeksan varianttia tunnistettiin translaatiottomilta alueilta. Suurin osa tunnistetuista varianteista, mukaan lukien useat lyhyet tandemtoistot (STR), sijoittuvat introneihin 2, 3 ja 4.
Olemme aiemmin kuvanneet SNCA:n intronissa 4 olevan erittäin polymorfisen CT-rikkaan alueen, jolla on neljä havaittua haplotyyppiä (Lutz ym., 2015). Vaikka tätä erittäin toistuvaa ja rakenteellisesti vaihtelevaa aluetta osoittautui vaikeaksi genotyypittää GATK4 HC:llä, pystyimme rakentamaan konsensussekvenssit kaikille 12 näytteelle ja havaitsimme kaikki neljä aiemmin havaittua haplotyyppiä (lisäkuva 2). Lisäksi tunnistimme intronissa 4 uuden STR:n, joka koostuu kolmen emäksen yksiköstä, joka toistuu 16 kertaa referenssissä. Tunnistimme 12 näytteestä kolme haplotyyppiä, joissa oli 9, 12 ja 15 kopiota TTG-toistoyksikköä. GATK HC genotyypitti oikein kaikki paitsi yhden PD-4:n haplotyypin, jonka kattavuus tällä alueella oli melko alhainen. Tämän näytteen annetuilla tiedoilla genotyyppi voidaan kuitenkin määrittää silmämääräisesti (taulukko 1).

TAULUKKO 1. Uusi tripletti-tandemtoisto intronissa 4 (chr4: 90713442).
Käytimme GATK HC:n havaitsemia lyhyitä variantteja yhdessä lukupohjaisen vaiheistustyökalun WhatsHapin (Martin et al., 2016) kanssa CCS-lukujen vaiheistamiseen koko lokuksen alueella, ja menestyksen vaihteluväli johtui pääasiassa heterotsygoottisen variantin tiheydestä koko lokuksen alueella. Näytteissä PD-1, PD-4, N-4, DLB-1 ja DLB-4 oli pitkiä matalan heterotsygotiittisyyden pätkiä, joissa oli hyvin vähän lyhyitä vaiheistuslohkoja, kun taas muissa näytteissä vaiheistuslohkot vaihtelivat 7-18-kertaisesti keskimääräiseen lukupituuteen, jopa 54 kb:n pituisiin lukulohkoihin asti (Täydentävä kuva 3).
Kohdennettu cDNA-kaappaus identifioi uusia alku- ja loppusijoituspaikkoja
Käsittelimme PacBion cDNA-tiedot (Iso-seq) PacBion SMRT-analyysiohjelmiston avulla. Kun Iso-Seq-data oli kartoitettu hg19:ään ja artefaktit oli poistettu (lisätaulukko 3, lisäkuva 4), saimme lopullisen 41 SNCA-isoformin joukon (kuva 3). Kaikissa lopullisissa isomuodoissa on kaikki kanoniset liitospaikat (GT-AG tai GC-AG), ja niiden tukena on yhteensä vähintään 20 täyspitkää lukua. Suurimmalla osalla isomuodoista (28:lla 41:stä) on kaikki kuusi eksonia, ja ne eroavat toisistaan vain vaihtoehtoisten 5′ aloituskohtien ja 3′ UTR:n pituuksien osalta. 3′ UTR:n pituudet vaihtelivat 300 ja 2,6 kb välillä. Erittäin vaihtelevan vaihtoehtoisen 5′-alkukohdan käyttö SNCA:ssa tunnetaan; vähemmän tunnettua on vaihteleva 3′ UTR-pituus, jota oli aiemmin tutkittu käyttämällä RNA-seq-dataa, joka ei ratkaissut täyspitkän isoformin rakenteita (Rhinn et al., 2012). Iso-Seq-tiedot osoittavat, että vaihteleva 3′ UTR-pituus näyttää olevan parina kaikkien mahdollisten 5′-alkusijoituskohtien kombinaatioiden kanssa ilman etuoikeutettua kytkeytymistä. Lähes mikään alku- ja loppupaikan vaihtelusta ei muuta ennustettua avointa lukukehystä (Täydentävä kuva 5), ja sen ennustetaan kääntyvän kanoniseen 141 aminohapon sekvenssiin.

Kuva 3. Kohdistetun Iso-Seqin avulla kaapatut SNCA-isoformit tunnistavat uusia alku- ja loppukohtia. Suurin osa isoformien monimutkaisuudesta on peräisin vaihtoehtoisten 3′ UTR-pituuksien ja eksonin 1 yhdistelmäkäytöstä, ja muutama harvinainen vaihtoehtoinen liitospaikka löytyy eksonista 1 (vihreä), 2 (punainen) ja 4 (sininen). Kaikissa liitoskohdissa on kanoniset liitospaikat. Tunnistimme viisi isomuotoa, jotka ohittivat eksoni 5, ja kaksi isomuotoa, jotka ohittivat eksoni 3. Tunnistimme myös uusia aloitus- (oranssi) ja lopetuskohtia (violetti) intronissa 4. Kutsutut SNP:t on merkitty violetilla.
Validoimme lisäksi uudet (mutta kanoniset) liitoskohdat käyttämällä julkisesti saatavilla olevia lyhyitä luettuja liitosdatoja. Intropolis (v1, https://github.com/nellore/intropolis) -tietokanta yhdistää yli 21 000 julkisesti saatavilla olevaa RNA-seq. Koska vain yhdellä lyhyellä lukukerralla tuettujen liitosdatojen määrä on suuri, tarvitsemme tässä tutkimuksessa vähintään 10 lyhyen lukukerran tuen (yhdistettynä kaikista >21 000 RNA-seq-tietokannoista) vahvistaaksemme Iso-Seq-uutuusliitoksemme. Lukuun ottamatta PB.1016.253:n ja PB.1016.296:n (kuva 3) uusia risteymiä, kaikki muut uudet risteymät saavat tukea Intropolis-datajoukosta. Mielenkiintoista on, että näillä uusilla yhteyksillä on huomattavasti vähemmän tukea lyhyille lukemille kuin Gencode-annotoiduilla yhteyksillä. Esimerkiksi PB.1016.139:n kahta uuden eksonin tuomaa uutta yhtymäkohtaa tukee 2 519 ja 44 Intropolis-lyhytlukulukua, kun taas neljää muuta tunnettua yhtymäkohtaa tukee yli miljoona lyhyttä lukulukua. Tämä osoittaa, että kohdennetulla rikastamisella, jossa käytetään koko transkriptomin pituuden sekvensointia, voidaan havaita harvinaisia, uusia isoformeja.
Havaitsimme kaksi isoformia, joissa eksoni 3 on hylätty (SNCA126), ja viisi isoformia, joissa eksoni 5 on hylätty (SNCA112). Tässäkin tapauksessa näiden kahden eksonin skippausryhmän splikointidiversiteetti johtuu enimmäkseen vaihtoehtoisten 5′ aloituskohtien erilaisesta käytöstä ja vaihtelevasta 3′ UTR:n pituudesta. ORF-ennuste osoittaa, että eksoni 3:n tai eksoni 5:n skippaaminen lyhentää ORF:ää mutta säilyttää lukukehyksen. Kolmella isomuodolla on intronissa 4 sijaitsevia uusia 3′-päätepaikkoja. ORF-ennuste osoittaa, että tämä johtaa typistettyyn proteiinituotteeseen.
Tunnistimme aiemmin noteeraamattoman 5′-alkukohdan, joka sijaitsee intronissa 4 (hg19 chr4: 90692548-90693045, kuva 3). Tähän uuteen starttiin liittyvät kolme isoformia koostuvat uudesta starttikohdasta, eksonista 5 ja vaihtelevista 3′ UTR-pituuksista. Mielenkiintoista on, että vaikka GTExistä ja Sandor et al. (2017) julkisesti ladatut lyhyet lukutiedot ja CAGE-piikkitiedot (FANTOM5) eivät tukeneet tätä uutta aloituskohtaa, äskettäinen julkinen NA12878:n suora RNA-tietoaineisto4 sisälsi vain yhden SNCA-transkriptin, joka vahvisti tämän vaihtoehtoisen aloituskohdan. Lisäksi Intropolis short read junction -tiedot vahvistavat eksoni 5:n ja uuden aloituskohdan välisen uuden liitoskohdan. Mielenkiintoista on, että tämän uudenlaisen 5′-aloituskohdan ennustetaan tuovan uusia peptidejä säilyttäen samalla lukukehyksen eksonissa 5.
Tunnistimme myös kolme SNCA-transkriptiä, joilla on uudet lopetuskohdat (kuva 3). Kaksi isoformia (PB.1016.383, PB.1016.384) käytti pidennettyä 3′ UTR:ää eksonissa 4, kun taas kolmas isoformi (PB.1016.381) käytti uutta 3′ eksonia intronissa 4. Uuden viimeisen eksonin ja edellisen eksonin väliset uudenlaiset yhtymäkohdat saavat tukea julkisista lyhyen lukemisen yhtymäkohtia koskevista tiedoista (Intropolis). Uudet 3′ UTR:t johtavat typistettyyn ORF-ennusteeseen.
Käyttämällä normalisoitua kokopituuslukujen lukumäärää isomuodon runsauden vertailuarvona havaitsimme, että yksi SNCA:n kanonisista isomuodoista (PB.1016.131) on runsain, ja sen esiintyvyys on 50-60 % kaikissa koehenkilönäytteissä (lisätaulukko 4). Ryhmittelimme 41 isoformia edelleen niiden splikointimallin mukaan (taulukko 2). Isomuotojen, joissa on kaikki kuusi eksonia, osuus runsaudesta on 95-97 %. Aiemmat tutkimukset ovat osoittaneet, että eksoni 3 puuttuvien isoformien (SNCA126) ilmentyminen on lisääntynyt huomattavasti DLB-näytteiden otsalohkossa verrattuna normaaliin (Beyer ym., 2008); aggregoidut isoformien lukumäärämme osoittavat, että kolmessa DLB-näytteessä on hieman kohonnut lukumäärätaso verrattuna normaalinäytteisiin, samoin kuin SNCA112 (eksoni 5:n skippaus) -muunnokset PD- ja DLB-näytteissä normaaleihin näytteisiin nähden.

Taulukko 2. Isoformien lukumäärä. SNCA-isoformien runsaus kussakin näytteessä, aggregoituna splikointimallin mukaan.
Full-Length cDNA Enables Isoform-Level Phasing Information
Kutsuimme SNP:t käyttämällä cDNA:ta kasaamalla kaikki täyspitkät lukemat 12:sta näytteestä varianttien kutsumiseksi (ks. kohta “Menetelmät”). Yhteensä kutsuttiin neljä SNP:tä, ja kaikki olivat aiemmin annotoituja dbSNP:ssä (taulukko 3, kuva 3). Kaikki neljä SNP:tä sijaitsevat ei-CDS-alueilla, yksi 3′ UTR:ssä (eksoni 6), yksi intronissa 4 ja kaksi 5′ UTR:ssä (eksoni 1). 3′ UTR:n SNP:n (chr4: 90646886) peittävät vain isomuodot, joiden 3′ UTR on vähintään ~1 kb pitkä, ja näin ollen kaikki kanoniset isomuodot eivät kata tätä SNP:tä. Intronin 4 SNP (chr4: 90743331) kattavat vain uudet vaihtoehtoiset 3′-loppuiset isomuodot (PB.1016.383, PB.1016.384), eikä se liity mihinkään muuhun SNP:hen. Kaksi 5′ UTR:n SNP:tä (chr4: 90757312 ja chr4: 90758389) katetaan kahdella toisensa poissulkevalla eksoni 1 -käytöllä, joten ne eivät myöskään liity toisiinsa.

Taulukko 3. cDNA:n SNP-tiedot.
Tämänhetkinen toimintatapamme rajoittuu siihen, että pystymme soittamaan vain korvautumismuunnoksia transkriboiduilla alueilla, joilla on riittävä peitto. Verrattaessa SNP-luetteloamme hg19 dbSNP-annotaatioon käy ilmi, että suurin osa väliin jääneistä SNP:istä tai varianteista oli joko alle 1 %:n frekvenssillä populaatiossa, ne eivät olleet yksittäisiä nukleotidisubstituutioita tai ne sijaitsivat matalan kompleksisuuden alueiden vieressä. Esimerkiksi rs77964369:n (chr4: 90646532) ilmoitetaan olevan 50/50-tiheydeltään T/A; tämä T on kuitenkin vierekkäin 11 genomisen As-osan kanssa alavirtaan. Manuaalinen tarkastelu Iso-Seq-lukupinosta, jossa on ~1 300 lukua tässä kohdassa, ei viittaa todisteisiin variaatiosta ainakaan 12 näytteemme joukossa.
Näytekohtaisten lukujen avulla kutsumme kunkin näytteen genotyypin kussakin SNP-paikassa (taulukko 3). Sen lisäksi, että PD-2:lla on liian vähän lukemia ja se on epäselvä kaikkien neljän SNP:n osalta, pystyimme kutsumaan genotyypin useimmille muille näytteille. Erityisesti DLB-3 oli ainoa näyte, joka on heterotsygoottinen kaikilla SNP-paikoilla. Muuten emme havainneet mitään tilakohtaista mallia, jossa suosittaisiin yhtä genotyyppiä muihin nähden.
Keskustelu
Kuvaamme ensimmäisen tutkimuksen, jossa käytettiin SNCA-geenin kohdennettua rikastamista multipleksoiduissa gDNA- ja cDNA-kirjastoissa neurologisten sairauksien tutkimiseen pitkän lukusekvensoinnin avulla. PacBio Sequel -järjestelmän pitkät lukupituudet helpottivat SNCA-geenin koko transkriptin pituisen isoformirepertuaarin sekvensointia. Paljastimme monimuotoisuuden vaihtoehtoisten 5′ aloituskohtien ja vaihtelevien 3′ UTR-pituuksien käytössä ja havaitsimme tunnettuja eksonien skippaustapahtumia, kuten eksoni 3:n deletion (SNCA126) ja eksoni 5:n deletion (SNCA112). Lisäksi suuren intronin 4 sisällä tunnistettiin uusia vaihtoehtoisia aloitus- ja lopetuskohtia, joiden ennustetaan kääntyvän uusiksi proteiineiksi. On todennäköistä, että kohdennetun kaappauksen suuri sekvensointipeiton syvyys yhdessä täydellisten transkriptien sekvensointikyvyn kanssa mahdollisti näiden aiemmin kuvaamattomien isoformien havaitsemisen.
SNCA-proteiinin eri isoformien biologista ja patologista merkitystä ei ole vielä täysin selvitetty. Spesifiset SNCA:n translaation jälkeisen modifikaation ja splikoinnin isoformit on kuitenkin yhdistetty solunsisäisiin aggregaatioalttiuksiin (Kalivendi ym., 2010), ja ne ilmentyvät eri tavoin ihmisen synukleinopatioissa (Beyer ym., 2008; Beyer ja Ariza, 2012). SNCA:n translaation jälkeistä modifikaatiota koskevat tutkimukset osoittivat, että Lewyn kappaleet, synukleinopatioiden patologinen tunnusmerkki, sisältävät runsaasti fosforyloitua, nitroitua ja monoubikvitinoitua SNCA:ta (Kim ym., 2014). Myös transkription jälkeisen modifikaation vaikutuksia SNCA:n aggregaatioon on tutkittu. Vaihtoehtoisen splikoinnin on ehdotettu vaikuttavan SNCA-aggregaatioon. Joko eksoni 3:n tai 5:n poisto ennustaa toiminnallisia seurauksia: kun taas eksoni 3:n poisto (SNCA126) johtaa N-terminaalisen proteiinin ja membraanin vuorovaikutusdomeenin katkeamiseen, mikä voi johtaa vähäisempään aggregaatioon, ja eksoni 5:n poisto (SNCA112) voi johtaa lisääntyneeseen aggregaatioon, mikä johtuu jäsentymättömän C-terminaalin huomattavasta lyhenemisestä (Lee et al., 2001; Beyer, 2006). DLB-potilaiden otsalohkossa SNCA112:n määrä lisääntyy selvästi kontrolleihin verrattuna (Beyer ym., 2008), kun taas SNCA126:n määrä vähenee DLB-potilaiden prefrontaalisessa aivokuoressa (Beyer ym., 2006). Sitä vastoin SNCA126-ekspressio osoitti lisääntyneen PD-aivojen frontaalisella aivokuorella eikä merkittäviä eroja MSA:ssa (Beyer ym., 2008). SNCA98 on aivospesifinen splice-variantti, josta puuttuu sekä eksoni 3 että 5 ja jolla on erilaiset ekspressiotasot sikiön ja aikuisen aivojen eri alueilla. SNCA98:n yliekspressiota on raportoitu DLB:n, PD:n (Beyer ym., 2007) ja MSA:n (Beyer ym., 2008) otsalohkoissa verrattuna kontrolleihin. Lisäksi transkription jälkeisellä prosessilla, joka johtaa vaihtoehtoiseen 3′UTR-käyttöön, on raportoitu olevan vaikutuksia mRNA:n vakauteen ja lokalisaatioon (Fabian et ai., 2010; Rhinn et ai., 2012; Yeh ja Yong, 2016). Lisätutkimukset eri tunnettujen SNCA-proteiinin isoformien aggregaatioalttiudesta ja Lewyn kappaleiden koostumuksesta ovat perusteltuja. Lisäksi tutkimuksemme loi pohjan aiemmin tunnettujen ja uusien transkriptien mRNA-kvantifiointianalyyseille suuremmassa otoskokoluokassa, joka koostui koehenkilöistä, joilla on erilaisia kliinispatologisia vaiheita, käyttäen useita aivoalueita kustakin koehenkilöstä. Nämä analyysit SNCA:n aivoaluekohtaisesta transkriptomimaisemasta neuropatologisen vakavuuden yhteydessä ovat informatiivisia sen suhteen, mikä on tiettyjen SNCA:n transkriptioiden isoformien rooli neuropatologisten vaiheiden etenemisessä ja Lewyn kappaleiden ja Lewyn neuriittien tiheyden vakavuudessa.
Tässä artikkelissa keskityimme luomaan sekvensointi- ja analyysistandardin samoista koehenkilöistä tuotettujen kohdennettujen gDNA:n ja cdna:n aineistojen analysoimiseksi. Tämä on tehokas lähestymistapa, joka mahdollisesti mahdollistaa gDNA-sekvenssien vaiheistamisen tietyn geenin koko alueella täyspitkän transkriptin isoformien sekvenssin heterotsygotiuden perusteella. Tämän tutkimuksen PacBio-kohdennetut gDNA-tiedot tuottivat vaiheistettuja lohkoja, jotka kattoivat 81 prosenttia 114 kb:n alueesta, jonka keskipisteenä oli SNCA, ja pisin vaiheistettu lohko oli yli 54 kb. Koska gDNA:n vaiheistusta rajoittavat lukupituus ja heterotsygoottius, lukupituuden kasvattaminen tuottaa todennäköisesti suurempia vaiheistettuja lohkoja.
gDNA:n varianttianalyysi vahvisti tunnetut ja tunnisti uusia lyhyitä tandemtoistoja (STR) intronisilla alueilla. Esimerkiksi aiemmin löysimme kloonauksen ja Sanger-sekvensoinnin avulla vaiheistetun sekvensoinnin avulla neljä erillistä haplotyyppiä intronisella CT-rikkaalla alueella, joka koostui vaihtelevien toistuvien sekvenssien klusterista (Lutz ym., 2015). Osoitimme, että tietty haplotyyppi, jota kutsutaan haplotyypiksi 3, aiheutti riskin sairastua Lewyn kappaleen patologiaan Alzheimer-potilailla. Tässä validoimme tämän erittäin polymorfisen matalan kompleksisuuden alueen sekvenssin ja sen neljä määriteltyä haplotyyppiä. Vaikka otoskoko oli pieni, haplotyyppiä 3 esiintyi yksinomaan tautipotilailla (yksi PD-potilas, kaksi DLB-potilasta), mikä vastaa aiempia havaintojamme. Pilottitulokset ja aiempi julkaisumme antavat lähtökohdan toistaa synukleiinisairauksien assosiaatioanalyysit tarkasti, eli pitkien lukukertojen avulla, määriteltyjen STR:ien ja rakenteellisten haplotyyppien kanssa käyttäen suurempaa otoskokoa.
Kirjoituksessamme osoitimme PacBio Sequel -järjestelmän kyvyn löytää uusia kokopitkiä transkriptejä ja luonnehtia tautiin osallisena olevan geenin täydellistä kokopituista transkriptioiden repertuaaria. Lisäksi osoitimme, että gDNA:n pitkät lukemat määrittelevät tarkemmin lyhyet rakenteelliset variantit ja haplotyypit, mukaan lukien STR:t, ja voivat siten helpottaa muiden sairauteen liittyvien varianttien kuin SNP:iden löytämistä ja validointia. Kokonaisuutena tämä uusi tieto on erittäin arvokasta ja sovellettavissa edistettäessä ymmärrystämme geneettisestä etiologiasta, johon voi liittyä häiriöitä transkriptiomaisemassa ja joka on monimutkaisten ihmissairauksien taustalla, mukaan lukien ikään liittyvät hermoston rappeutumissairaudet, kuten synukleinopatiat.
Datan saatavuus
Kolmen SMRT-solun gDNA:n raakadatatat ovat saatavissa osoitteessa Zenodo.org osoitteessa, jossa on osoitteessa doi: 10.5281/zenodo.1560688. Yksi cDNA-raakadatan SMRT-solu on saatavilla osoitteessa Zenodo.org, doi: 10.5281/zenodo.1581809. Käsitellyt gDNA- ja cDNA-tulokset, mukaan lukien gDNA-variantit ja cDNA-isoformit, ovat saatavilla osoitteessa Zenodo.org, doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F osallistui tutkimuksen ideointiin ja suunnitteluun. ET ja WR järjestivät sekvenssitietokannat, suorittivat sekvenssianalyysit ja valmistelivat kaikki kuvat ja taulukot. O-CG ja JB käsittelivät aivokudokset ja nukleiininäytteiden valmistelut. TH tuotti sekvensointiaineistot. SK suunnitteli ja hankki reagenssit. OC-F, ET ja WR kirjoittivat käsikirjoituksen ensimmäisen luonnoksen. OC-F hankki rahoituksen. Kaikki kirjoittajat osallistuivat käsikirjoituksen valmisteluun, lukivat ja hyväksyivät toimitetun version.
Rahoitus
Tämän työn rahoitti osittain National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS).
Eriintyneisyysristiriitoja koskeva lausunto
ET, WR, TH ja SK työskentelevät tai työskentelivät Pacific Biosciencesin työntekijöinä tutkimuksen aikana.
Muut kirjoittajat ilmoittavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, jotka voitaisiin tulkita mahdolliseksi eturistiriidaksi.
Kiitokset
Tämä käsikirjoitus on julkaistu ennakkopainoksena BioRxivissä (Tseng ym, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Lisäaineisto
Tämän artikkelin lisäaineisto löytyy verkosta osoitteesta: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizonan tutkimus ikääntymisestä ja hermoston rappeutumishäiriöistä sekä aivojen ja kehon luovutusohjelma. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-synukleiinin rakenne, posttranslationaalinen modifikaatio ja vaihtoehtoinen pilkkominen aggregaation tehostajina. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Alfa-synukleiinin posttranslationaalinen modifikaatio ja vaihtoehtoinen splikointi neurodegeneraation laukaisijana. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I. ja Ariza, A. (2008). Alfa-synukleiinin, parkiinin ja synfiliini-1-isoformien erilainen ilmentyminen Lewyn kappale -taudissa. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Uuden alfa-synukleiinin isomuodon tunnistaminen ja karakterisointi sekä sen rooli Lewyn kappale -sairauksissa. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Alhaiset alfa-synukleiini 126:n mRNA-tasot Lewyn kappaleiden dementiassa ja Alzheimerin taudissa. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N. ja Filipowicz, W. (2010). MikroRNA:iden säätely mRNA:n translaatiosta ja stabiilisuudesta. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Hapettimet indusoivat Α-synukleiinin vaihtoehtoista splikointia: vaikutukset Parkinsonin tautiin. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., ja Halliday, G. M. (2014). Alfa-synukleiinin biologia Lewyn kappale -sairauksissa. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgeenireseptorivariantti AR-V9 ekspressoituu yhdessä AR-V7:n kanssa eturauhassyövän etäpesäkkeissä ja ennustaa abirateroniresistenssiä. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C. ja Lee, S. J. (2001). Kalvoon sitoutuneella Α-synukleiinilla on suuri aggregaatioalttius ja kyky siementää sytosolisen muodon aggregaatiota. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., and Chiba-Falek, O. (2015). Sytosiini-tymiini (CT)-rikas haplotyyppi SNCA:n intronissa 4 antaa riskin Lewyn kappaleen patologialle Alzheimerin taudissa ja vaikuttaa SNCA:n ilmentymiseen. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., ym. et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., ja Perry, R. H. (1999). Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on dementia with Lewy bodies. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M. ja Isacson, O. (2012). Täyspitkän alfa-synukleiinin ja sen kolmen vaihtoehtoisesti splikoidun muunnoksen transkriptioiden ilmentymistasot Parkinsonin taudin aivoalueilla ja transgeenisessä hiirimallissa, jossa alfa-synukleiini yli-ilmentyy. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to ten of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Vaihtoehtoinen Α-synukleiinin transkriptien käyttö konvergenttina mekanismina Parkinsonin taudin patologiassa. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Puhdistettujen potilasperäisten dopamiinineuronien transkriptominen profilointi tunnistaa yhtenevät häiriöt ja Parkinsonin taudin terapeuttiset hoidot. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R. ja Südhof, T. C. (2014). Neureksiinin vaihtoehtoisen pilkkomisen kartografia, joka on kartoitettu yhden molekyylin pitkän lukukerran mRNA-sekvensoinnilla. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). FMR1-spilsaatiomuunnosten maiseman muuttunut ilmentyminen premutaatiokantajilla. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). SNCA-geenin ja Parkinsonismin välinen yhteys. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). MRNA:iden vaihtoehtoinen polyadenylaatio: 3′-kääntämättömällä alueella on merkitystä geeniekspressiossa. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar