ChIP-seq är en fantastisk teknik som gör det möjligt för oss att undersöka de fysiska bindningsinteraktionerna mellan protein och DNA med hjälp av nästa generations sekvensering. I den här artikeln ger jag en kort genomgång av ChIP och introducerar sekvenseringstekniken för kromatinimmunutfällning (ChIP-seq), som kombinerar ChIP med nästa generations sekvensering.
Vad är kromatinimmunutfällning?
Kromatinimmunutfällning (ChIP) gör det möjligt för oss att bestämma proteinbindningsställen på DNA. Kromatin är det komplex av DNA som är paketerat med histonproteiner i nukleosomer. ChIP utnyttjar reversibla tvärbindningar som skapas mellan DNA och associerade proteiner genom formaldehydfixering av celler eller vävnad. Det fixerade kromatinet skäras fysiskt och DNA-fragment som är associerade med ett visst protein immunutskiljs och analyseras selektivt. Analysen kan ske lokus för lokus med hjälp av PCR, men vanligare är att ChIP undersöks med mikroarrayer (ChIP-chip) eller nästa generations sekvensering (ChIP-seq).
Hur fungerar ChIP-chip?
ChIP-on-chip, eller ChIP-chip, kombinerar kromatinimmunutfällning med mikroarray-analys (chip). År 2001 publicerade Jason Lieb i Pat Browns labb vid Stanford den första ChIP-chip-artikeln (Lieb 2001). I denna metod appliceras de DNA-fragment som fälls ut med ett visst protein på ett mikroarray-chip för analys. Detta genererar en global bild av var proteinet binder, till skillnad från att helt enkelt förhöra enskilda bindningsställen genom PCR. Även om detta var ett revolutionerande tillvägagångssätt är tekniken begränsad av de arrayer som används. För det första behöver man ett mikroarray för det genom man vill studera, och i början och mitten av 2000-talet var detta ofta inte fallet. För det andra begränsas dessa mikroarrayer av kvaliteten på referensgenomet och förmågan att utforma prober som fungerar på en array. För det tredje är arrayer begränsade när det gäller hur stor del av genomet som kan representeras, och de är vanligtvis uppdelade över hela genomet med en upplösning på 100 eller 1000 bp. Det finns också frågor om bias vid amplifiering av ChIP-dna-fragmenten, normalisering av array-data och jämförbarhet mellan array-plattformar.
Hur fungerar ChIP-seq?
Chromatin immunoprecipitation sequencing, eller ChIP-seq, kombinerar ChIP med nästa generations sekvensering (Barski 2007, Johnson 2007). ChIP-seq-protokoll har anpassats från ChIP-chip-metoder: proteiner tvärbinds till sitt bundna DNA genom formaldehydbehandling, celler homogeniseras och kromatin skjuvas och immunprecipiteras med antikroppsbundna magnetpärlor. Det immunoprecipiterade DNA:t används sedan som indata för ett protokoll för att förbereda nästa generations sekvenseringsbibliotek, där det sekvenseras och analyseras med avseende på DNA-bindningsställen. Se figuren nedan för en sammanfattning av ChIP-seq-arbetsflödet och ett exempel på ChIP-seq-resultat (återgivet med vänligt tillstånd från Dominic Schmidt (Schmidt 2009)).
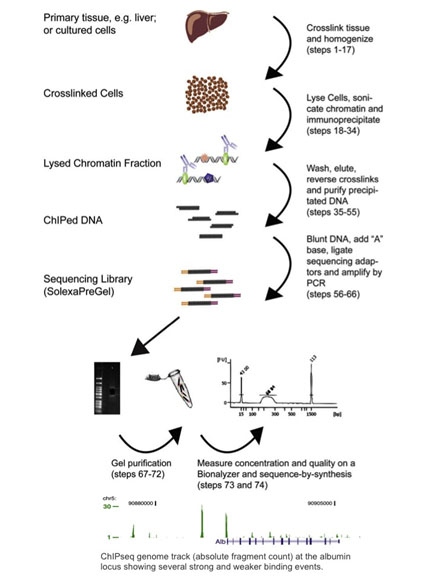
Och även om majoriteten av de cirka 400 artiklar som publicerats hittills har analyserats på Illumina-plattformen, kan ChIP-seq utföras på vilken nästa generations sekvenserare som helst (Wold 2008). ChIP-seq har fått stor spridning sedan det först rapporterades 2007. Faktum är att det nästan helt har ersatt ChIP-Chip, eftersom det möjliggör genomomfattande analyser och inte har de begränsningar som diskuterats ovan.
Vad kan man egentligen göra med ChIP-seq
ChIP-seq är ett kraftfullt och mångsidigt verktyg, och det finns många bra exempel på användning av ChIP-seq i litteraturen. Jag har valt ut ett par av mina favoriter (från arbete som utförts i den kärnanläggning som jag förvaltar) för att illustrera vad som är möjligt, och har inkluderat exempel där ChIP-seq har inspirerat till utveckling av nya metoder:
- Antoni Hurtado, et al. utförde knock-down av FoxA1-“pionjärfaktorn”, vilket resulterade i minskad bindning av östrogenreceptorn (ER) vid mer än 50 % av kända ER-bindningsplatser. De visade att FoxA1 är en viktig regulator av ER-medierad transkription, vilket tyder på att den kan vara ett nytt och viktigt terapeutiskt mål för bröstcancer (Hurtado 2011).
- Dominic Shmidt, et al. använde ChIP-seq för att undersöka evolutionen av transkriptionsfaktorbindning. De fokuserade på CEBPA- och HNF4-bindning i levervävnad från fem ryggradsdjursarter: människa, mus, hund, opossum och kyckling. ChIP-chip skulle ha varit nästan omöjligt med tanke på de olika arter som var inblandade och komplexiteten i utformningen av prober (Schmidt 2010).
Fortsatta modifieringar av ChIP-seq-metoden har lett till flera nya metoder, bland annat ChIP-seq för analys av RNA-proteininteraktion samt DNase-seq och FAIRE-seq, som båda används för att identifiera regulatoriska regioner i DNA.
Sammanfattningsvis är ChIP-seq en mogen teknik som är att föredra framför ChIP-chip och som kan användas av nästan alla grupper som är intresserade av analys av DNA:proteininteraktion. Hur kan du använda ChIP-seq i din forskning?
Barski et al, High-resolution profiling of histone methylations in the human genome. Cell 129 (2007).
Hurtado et al, FOXA1 is a key determinant of estrogen receptor function and endocrine response.Nature Genetics (2011).
Johnson et al, Genome-wide mapping of in vivo protein-DNA interactions. Science (2007).
Lee et al, Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc (2006).
Lieb et al, Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001.
Morozova & Marra, Applications of next-generation sequencing technologies in functional genomics. Genomics (2008).
Schmidt et al, ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods (2009).
Schmidt et al, Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding Science (2010).
Wold & Myers, Sequence census methods for functional genomics. Nat. Methods (2008).
Har detta hjälpt dig? Dela gärna med dig till ditt nätverk.
.