- Introduktion
- Material och metoder
- Studieprover
- Genomiskt DNA och RNA-extraktioner
- Biblioteksberedning och sekvensering
- GDNA-fångst med hjälp av IDT Xgen® Lockdown®-prober och sekvensering av enstaka molekyler
- cDNA-fångst med hjälp av IDT Xgen® Lockdown®-prober och isoformsekvensering av enstaka molekyler (Iso-Seq)
- gDNA-analys
- Short Variant Analysis and Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Isoformanalys
- Isoform SNP Calling
- Resultat
- Targeted gDNA Capture Identified Known and Novel Variations
- Målinriktad cDNA-fångst identifierade nya start- och slutställen
- Fullängds cDNA möjliggör information om fasning på isoformnivå
- Diskussion
- Datatillgänglighet
- Författarnas bidrag
- Finansiering
- Intressekonfliktutlåtande
- Acknowledgments
- Supplementary Material
Introduktion
Transkriptionella och posttranskriptionella program kontrollerar genuttrycksnivåerna och/eller produktionen av flera olika mRNA-isoformer, och förändringar i dessa mekanismer resulterar i dysreglering av genuttrycket och differentiella uttrycksprofiler. Felaktig transkriptionell och posttranskriptionell genreglering är vanligt förekommande i vävnader i det mänskliga nervsystemet och bidrar till fenotypiska skillnader inom och mellan individer vid hälsa och sjukdom.
Dysreglering av alfa-synuklein-uttrycket har involverats i patogenesen för synukleinopatier, i synnerhet Parkinsons sjukdom (PD) och demens med Lewy-kroppar och (DLB). Även om rollen av SNCA-överuttryck i synukleinopatier, främst PD, är väletablerad, har vi här fokuserat på att fastställa den fullständiga repertoaren av SNCA-transkriptets isoformer i olika synukleinopatier. Tidigare har flera olika SNCA-transkriptisoformer beskrivits för SNCA-genen, som uppkommit genom alternativ splicing, transkriptionella startplatser (TSS) och val av polyadenyleringsställen (McLean et al., 2012; Xu et al., 2014). Alternativ splicing av de kodande exonerna ger upphov till SNCA 140, SNCA 112, SNCA 126 och SNCA 98, vilket resulterar i fyra proteinisoformer (Beyer och Ariza, 2012). Alternativa TSS i SNCA-genen resulterar i fyra olika 5′UTR, och alternativt val av olika polyadenyleringsställen bestämmer tre huvudlängder av 3′UTR, utan inverkan på proteinproduktens sammansättning (Beyer och Ariza, 2012). Vårt övergripande mål är att få nya insikter om bidraget från de olika SNCA mRNA-arter, kända och nya, till patogenesen och heterogeniteten hos synukleinopatier.
Hittills har de flesta studier använt sig av kortläsningssekvenseringsteknik för att undersöka transkriptomkomplexiteten i mänskliga hjärnor. Tillgången till tredje generationens teknik för långa avläsningar ger en oöverträffad och nästan fullständig bild av isoformstrukturer. Befintlig långläst transkriptsekvensering för mänskliga sjukdomsgener har dock använt en amplikonbaserad metod (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Även om detta tillvägagångssätt har varit framgångsrikt när det gäller att identifiera komplex alternativ splicing i mänskliga sjukdomsgener, är det begränsat till PCR-primerdesignen och kommer inte att avslöja alternativa start- och slutplatser. Riktad anrikning, t.ex. genom användning av IDT-sonder, kan ge en omfattande isoformvy av gener av intresse till en låg sekvenseringskostnad. Dessutom möjliggör mycket exakta transkriptläsningar i full längd isoformspecifik haplotypning.
Här presenterar vi den första kända studien med riktad fångst av gDNA och cDNA från SNCA-genregionen med hjälp av PacBio SMRT-sekvensering. SNCA-genregionen är ~114 kb lång och består av sex exoner med transkriptlängder på omkring 3 kb. Vi multiplexade 12 mänskliga hjärnprover från PD, DLB och normala kontrollprover och sekvenserade gDNA- och cDNA-biblioteket på PacBio Sequel-systemet. Vi beskriver de bioinformatiska analyser som användes för att identifiera SNPs, indels och korta tandemrepeats för gDNA-fångsten och haplotypning på isoformnivå för cDNA-data. Vi visar att riktad fångst är ett kostnadseffektivt sätt att gemensamt studera genomisk variation och alternativ splicing i en sjukdomsrelaterad neural gen.
Material och metoder
Studieprover
Studiekohorten (N = 12) bestod av individer med tre obduktionsbekräftade neuropatologiska diagnoser: (1) PD (N = 4), (2) DLB (N = 4) och (3) kliniskt och neuropatologiskt normala personer (N = 4). Vävnader från frontalkortexhjärnan erhölls genom Kathleen Price Bryan Brain Bank (KPBBB) vid Duke University, Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015) och Layton Aging and Alzheimer’s Disease Center vid Oregon Health and Science University. Neuropatologiska fenotyper fastställdes vid postmortalundersökning enligt väletablerade standardmetoder enligt metod och rekommendationer för klinisk praxis av McKeith och kollegor (McKeith et al., 1999, 2005). Tätheten av LB-patologin (i en standarduppsättning av hjärnregioner) fick poäng av mild, måttlig, allvarlig och mycket allvarlig. Studieproverna inom varje diagnosgrupp, PD och DLB, valdes noggrant ut så att svårighetsgraden av de klinisk-patologiska fenotyperna var liknande inom varje patologi. Alla hjärnor uppvisade hjärnstam, limbiska och neokortikala Lewy bodies (LBs), medan PD uppvisade svåra till mycket svåra McKeith-poäng i sub-nigra och amygdala. Alla hjärnor tyder inte på någon AD enligt CERAD-kriterierna och Braak och Braak-stadiet = II. De neurologiskt friska hjärnproverna togs från postmortala vävnader från kliniskt normala personer som undersöktes, i de flesta fall, inom ett år efter dödsfallet och befanns inte ha någon kognitiv störning eller parkinsonism och neuropatologiska fynd som var otillräckliga för att diagnostisera PD, Alzheimers sjukdom (AD) eller andra neurodegenerativa sjukdomar. Alla prover var vita. Demografiska uppgifter och neuropatologi för dessa försökspersoner sammanfattas i kompletterande tabell 1. Projektet godkändes av Duke Institution Review Board (IRB) som gav ett etiskt godkännande. Metoderna utfördes i enlighet med relevanta riktlinjer och förordningar.
Genomiskt DNA och RNA-extraktioner
Genomiskt DNA extraherades från hjärnvävnader med hjälp av standardprotokollet från Qiagen (Qiagen, Valencia, CA). Totalt RNA extraherades från hjärnprover (100 mg) med hjälp av TRIzol-reagens (Invitrogen, Carlsbad, CA) följt av rening med RNeasy-kit (Qiagen, Valencia, CA) enligt tillverkarens protokoll. gDNA- och RNA-koncentrationen bestämdes spektrofotometriskt, och RNA-provernas kvalitet och avsaknad av signifikant nedbrytning bekräftades genom mätningar av RNA-integritetssiffran (RIN, kompletterande tabell 1) med hjälp av en Agilent Bioanalyzer.
Biblioteksberedning och sekvensering
GDNA-fångst med hjälp av IDT Xgen® Lockdown®-prober och sekvensering av enstaka molekyler
Ungefär 2 μg av varje gDNA-prov klipptes till 6 kb med hjälp av Covaris g-TUBE och ligerades med streckkodade adaptrar. En ekvimolär pool av 12-plex barkodade gDNA-bibliotek (totalt 2 μg) matades in i den sondebaserade infångningen med en skräddarsydd SNCA-genpanel.
Ett SMRTBell-bibliotek konstruerades med hjälp av 626 ng av infångat och återamplifierat gDNA1.
cDNA-fångst med hjälp av IDT Xgen® Lockdown®-prober och isoformsekvensering av enstaka molekyler (Iso-Seq)
Omkring 100-150 ng totalt RNA per reaktion transkriberades omvänt med hjälp av Clontech SMARTer cDNA-synteskittet och 12 provspecifika streckkodade oligo dT (med PacBio 16mer streckkodssekvenser, se tilläggsmetoder). Tre reaktioner för omvänd transkription (RT) bearbetades parallellt för varje prov. PCR-optimering användes för att bestämma det optimala antalet amplifieringscykler för de storskaliga PCR-reaktionerna nedströms. En enda primer (primer IIA från Clontech SMARTer-kitet 5′ AAG CAG TGG TGG TAT CAA CGC AGA GTA C 3′) användes för alla PCR-reaktioner efter RT. Storskaliga PCR-produkter renades separat med 1X AMPure PB-beads, och bioanalysatorn användes för QC. En ekvimolär pool av 12-plex barcoded cDNA-bibliotek (totalt 1 μg) matades in i den sondbaserade infångningen med en skräddarsydd SNCA-genpanel.
Ett SMRTBell-bibliotek konstruerades med hjälp av 874 ng av infångat och återamplifierat cDNA2. En SMRT Cell 1M (6 h film) sekvenserades på PacBio Sequel-plattformen med hjälp av 2.0-kemi.
gDNA-analys
Sekvensering av de streckkodade gDNA-data kördes på tre SMRT Cells 1M med hjälp av 2.0-kemi. Data demultiplexades genom att köra programmet Demultiplex Barcodes i PacBio SMRT Link v6.0.
Short Variant Analysis and Phasing
Circular Consensus Sequence (CCS) reads genererades med hjälp av SMRT Analysis 6.0 från varje demultiplexad datauppsättning och anpassades till hg19-referensgenomet med minimap2. PCR-duplikat från amplifiering efter fångst identifierades genom kartläggning av slutpunkter och märktes med hjälp av ett anpassat skript. Korta varianter kallades med hjälp av GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Efter en första filtrering med hjälp av täckningsdjup och kvalitetsmetriker inspekterades varianter manuellt i IGV3. Om varianter inte fasade med närliggande SNPs filtrerades de manuellt. De variantplatser som klarade den manuella kureringen användes tillsammans med de deduplicerade CCS-anpassningarna för läsbackad fasning med WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Subsekvenser som anpassats till chr4: 90742331-90742559 (hg19) extraherades för varje prov. Efter att ha inspekterat storleksfördelningen för dessa undersekvenser klustrades de efter storlek och sekvenslikhet med hjälp av en kombination av python och MUSCLE (Edgar, 2004), och en konsensussekvens genererades oberoende av varandra för varje kluster.
Anpassade skript och arbetsflöden beskrivs vidare i https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Isoformanalys
Sekvensering av de streckkodade cDNA-data skedde på en SMRT Cell 1M på PacBio Sequel-systemet med hjälp av 2.0-kemi. Bioinformatisk analys gjordes med hjälp av programmet IsoSeq3 i PacBio SMRT Analysis v6.0.0.0 för att få högkvalitativa isoformsekvenser i full längd (se kompletterande metoder för mer information).
Isoform SNP Calling
Fullängdsavläsningar som var associerade med de slutliga 41 isoformerna från alla 12 proverna anpassades till hg19-genomet för att skapa en pileup. Baser med en QV på mindre än 13 exkluderades. Vid varje position med minst 40 basers täckning tillämpades sedan ett exakta Fisher-test med Bonferroni-korrigering med en p-gräns på 0,01. Endast substitutions-SNP som inte ligger nära homopolymerregioner (sträckor med 4 eller fler av samma nukleotid) har tagits fram. Efter SNP-kallandet fastställdes genotypen för varje prov genom att räkna antalet stödjande provspecifika fulllängdsavläsningar (FL) för varje prov. Om ett prov hade 5+ FL-reads som stödde både referens- och alternativbasen var det heterozygot. Om ett prov hade 5+ FL-reads som stödde en allel och färre än 5 FL-reads för den andra var det homozygot. I annat fall var det inte entydigt. Skripten finns tillgängliga på följande adress: https://github.com/Magdoll/cDNA_Cupcake.
Resultat
Vi utformade skräddarsydda prober för SNCA-genen och utförde riktad fångst av både gDNA och cDNA på ett multiplexat bibliotek som bestod av 12 mänskliga hjärnprover från PD, DLB och normala kontroller (figur 1, kompletterande tabell 1). GDNA- och cDNA-biblioteken sekvenserades på PacBio Sequel-plattformen. Bioinformatisk analys gjordes med hjälp av PacBio-programvaran följt av anpassad analys.
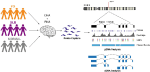
Figur 1. Schematisk presentation av studiens utformning. DNA- och RNA-material extraherades från postmortala hjärnvävnader från patienter med Parkinsons sjukdom, demens med Lewy Body och kontrollgrupper. gDNA- och cDNA-bibliotek gjordes med hjälp av sondhybridisering och sekvenserades på PacBio Sequel-systemet. Analysen utfördes med hjälp av PacBio-mjukvara och andra befintliga verktyg.
Targeted gDNA Capture Identified Known and Novel Variations
Efter att ha genererat cirkulära konsensussekvenser (CCS) och avlägsnat PCR-dubbletter (Supplemental Methods) erhöll vi 16- till 71-faldig genomsnittlig unik täckning av SNCA-genregionen. CCS-avläsningarna hade en genomsnittlig instickslängd på 2,9 kb och en genomsnittlig avläsningsnoggrannhet på 98,9 %. Med undantag för en region på 5 kb som avsiktligt inte täcktes av sonder på grund av förekomsten av LINE-element (hg19 chr4: 90697216-90702113) och en region på 2,1 kb med högt GC-innehåll runt exon 1, fanns det tillräcklig täckning för att genotypa båda haplotyperna för vart och ett av de 212 proverna (figur 2, kompletterande figur 1).

Figur 2. Målinriktad gDNA-fångst och fasning. Ett exempel som visar ett prov från varje tillstånd. Översta spåret visar en av SNCA-isoformerna, följt av gDNA-täckningen för de tre proverna. Variantspåret visar varje SNP och är färgkodat för heterozygot (lila), homozygot alternativ (orange) och homozygot referens (grå). Fasade block visas i ljusblått. Det nedre spåret visar var proberna placeras. Den bortfallande regionen i sondens utformning beror på två LINE-element i mitten av intron 4. För information om gDNA-täckning och fasindelning för alla 12 prover, se kompletterande figurer.
Med hjälp av GATK4 HC, kvalitetsbaserad filtrering och manuell kurering identifierade vi 282 SNPs och 35 indels, inklusive 8 SNPS och 13 indels som inte finns i dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (kompletterande tabell 2). Inga varianter identifierades i den kodande regionen för SNCA, även om åtta varianter identifierades i otranslaterade regioner. Majoriteten av de identifierade varianterna, inklusive flera korta tandemrepetitioner (STR), faller inom intron 2, 3 och 4.
Vi har tidigare beskrivit ett mycket polymorfiskt CT-rikt område i intron 4 av SNCA med fyra observerade haplotyper (Lutz et al., 2015). Även om denna mycket repetitiva och strukturellt variabla region visade sig vara svår att genotypa med GATK4 HC, kunde vi konstruera konsensussekvenser för alla 12 prover och observerade alla 4 av de tidigare upptäckta haplotyperna (kompletterande figur 2). Dessutom identifierade vi en ny STR i intron 4 som består av en trebasenhet som upprepas 16 gånger i referensen. I de 12 proverna identifierade vi tre haplotyper med 9, 12 och 15 kopior av TTG-upprepningsenheten. GATK HC genotypade alla dessa korrekt utom en haplotyp för PD-4, som hade ganska låg täckning i denna region. Med de givna uppgifterna för detta prov kan genotypen dock bestämmas genom visuell inspektion (tabell 1).

Tabell 1. En ny trippel tandemrepetition i intron 4 (chr4: 90713442).
Vi använde de korta varianter som upptäcktes av GATK HC tillsammans med det läsbaserade fasningsverktyget WhatsHap (Martin et al., 2016) för att fasa CCS-avläsningarna över locus, med en rad olika framgångar som främst drevs av den heterozygota varianttätheten över locus. Proverna PD-1, PD-4, N-4, DLB-1 och DLB-4 hade långa sträckor med låg heterozygositet, med mycket få, korta fasblock, medan de andra proverna gav fasblock som varierade från 7 till 18 gånger den genomsnittliga avläsningslängden, upp till 54 kb (kompletterande figur 3).
Målinriktad cDNA-fångst identifierade nya start- och slutställen
Vi bearbetade PacBio cDNA-data (Iso-Seq) med hjälp av programvaran PacBio SMRT Analysis. Efter kartläggning av Iso-Seq-data till hg19 och avlägsnande av artefakter (kompletterande tabell 3, kompletterande figur 4) fick vi en slutlig uppsättning av 41 SNCA-isoformer (figur 3). Alla slutliga isoformer har alla kanoniska skarvplatser (GT-AG eller GC-AG) och stöds av sammanlagt 20 eller fler fullängdsavläsningar. Majoriteten av isoformerna (28 av 41) har alla sex exoner och skiljer sig endast i användningen av alternativa 5′-startplatser och 3′ UTR-längder. 3′ UTR-längderna varierade mellan 300 och 2,6 kb. Användningen av mycket varierande alternativa 5′-startplatser i SNCA är känd; vad som är mindre känt är den varierande 3′ UTR-längden, som tidigare hade studerats med hjälp av RNA-seq-data som inte löste upp isoformstrukturer i full längd (Rhinn et al., 2012). Iso-Seq-data visar att den variabla 3′ UTR-längden verkar vara kopplad till alla möjliga kombinationer av 5′-startplatser utan någon preferentiell koppling. Nästan ingen av variabiliteten i start- och slutplats förändrar den förutspådda öppna läsramen (kompletterande figur 5) och förutspås översättas till den kanoniska 141 aminosyrasekvensen.

Figur 3. SNCA-isoformer som fångats med hjälp av riktad Iso-Seq identifierar nya start- och slutplatser. Majoriteten av isoformkomplexiteten kommer från kombinatorisk användning av alternativa 3′ UTR-längder och exon 1, med ett fåtal sällsynta alternativa skarvplatser i exon 1 (grönt), 2 (rött) och 4 (blått). Alla korsningar har kanoniska skarvplatser. Vi identifierade fem isoformer som hoppade över exon 5 och två isoformer som hoppade över exon 3. Vi identifierade också nya start- (orange) och slutplatser (lila) i intron 4. Kallade SNPs är markerade i lila.
Vi validerade ytterligare de nya (men kanoniska) förgreningarna med hjälp av offentligt tillgängliga kortlästa förgreningsdata. Databasen Intropolis (v1, https://github.com/nellore/intropolis) kombinerar över 21 000 offentligt tillgängliga RNA-seq. På grund av den stora volymen av korsningsdata som stöds av endast en enda kort läsning, kräver vi för den här studien minst 10 kortläsningsstöd (kombinerat från alla >21 000 RNA-seq-dataset) för att bekräfta våra Iso-Seq nya korsningar. Med undantag för de nya korsningarna för PB.1016.253 och PB.1016.296 (figur 3) stöds alla andra nya korsningar av Intropolis-databasen. Intressant nog har dessa nya korsningar betydligt mindre stöd för korta läsningar än de Gencode-annoterade korsningarna. Till exempel stöds de två nya korsningarna i PB.1016.139 som introduceras av det nya exonet av 2 519 respektive 44 Intropolis-kortläsningar, medan de andra fyra kända korsningarna stöds av över 1 miljon kortläsningar. Detta visar kraften hos riktad anrikning med hjälp av transkriptomsekvensering i full längd för att upptäcka sällsynta, nya isoformer.
Vi observerade två isoformer med exon 3 skipping (SNCA126) och fem isoformer med exon 5 skipping (SNCA112). Återigen kommer splicingdiversiteten i dessa två grupper som hoppar över exon främst från den varierande användningen av alternativa 5′-startplatser och varierande 3′ UTR-längd. ORF-prediktion visar att om exon 3 eller exon 5 skippas förkortas ORF:n men läsramen bibehålls. Tre isoformer har nya 3′-ändarplatser i intron 4. ORF-prediktion visar att detta resulterar i en trunkerad proteinprodukt.
Vi identifierade en tidigare oannoterad 5′-startplats i intron 4 (hg19 chr4: 90692548-90693045, figur 3). De tre isoformerna som är associerade med denna nya startplats består av den nya startplatsen, exon 5 och varierande 3′ UTR-längder. Intressant nog, medan offentligt nedladdade kortläsningsdata från GTEx och Sandor et al. (2017) och CAGE-toppdata (FANTOM5) inte stödde denna nya startplats, innehöll en nyligen publicerad NA12878 direkt RNA-datauppsättning4 endast ett SNCA-transkript som bekräftade denna alternativa startplats. Vidare bekräftas den nya korsningen mellan exon 5 och den nya startplatsen av Intropolis short read junction data. Intressant nog förutspås denna nya 5′-startplats introducera nya peptider samtidigt som läsramen i exon 5 bibehålls.
Vi identifierade också tre SNCA-transkript med nya slutplatser (figur 3). Två isoformer (PB.1016.383, PB.1016.384) använde en förlängd 3′ UTR i exon 4, medan den tredje isoformen (PB.1016.381) använde en ny 3′ exon i intron 4. De nya korsningarna mellan det nya sista exonet och det föregående exonet stöds av offentliga kortlästa korsningsdata (Intropolis). De nya 3′ UTR:erna resulterar i en trunkerad ORF-prediktion.
Om vi använder det normaliserade antalet läsningar i full längd som ett mått på förekomsten av isoformer finner vi att en av de kanoniska SNCA-isoformerna (PB.1016.131) är den mest frekventa, med en förekomst på 50-60 % i alla prover (kompletterande tabell 4). Vi grupperade vidare de 41 isoformerna efter deras splicingmönster (tabell 2). Isoformer som har alla sex exoner står för 95-97 % av abundansen. Tidigare studier har visat en markant ökning av uttrycket av isoformer som saknar exon 3 (SNCA126) i den frontala cortexen hos DLB-prover jämfört med normala prover (Beyer et al., 2008); våra aggregerade isoformsräkningar visar att tre av DLB-proverna har en något förhöjd räkningsnivå jämfört med de normala proverna samt SNCA112 (exon 5 skipping) varianter för PD och DLB jämfört med normala prover.

Tabell 2. SNCA isoforms abundans för varje prov, aggregerat efter splicingmönster.
Fullängds cDNA möjliggör information om fasning på isoformnivå
Vi kallade SNP:er med hjälp av cDNA genom att stapla alla fullängdsavläsningar från de 12 proverna för att kalla varianter (se avsnittet “Metoder”). Totalt fyra SNP kallades och alla var tidigare annoterade i dbSNP (tabell 3, figur 3). De fyra SNP:erna är alla belägna i icke-CDS-regioner, en i 3′ UTR (exon 6), en i intron 4 och två i 5′ UTR (exon 1). SNP:en i 3′ UTR (chr4: 90646886) täcks endast av isoformer med en 3′ UTR som är minst ~1 kb lång, och därför täcker inte alla kanoniska isoformer denna SNP. SNP:n för intron 4 (chr4: 90743331) täcks endast av de nya alternativa 3′-ändliga isoformerna (PB.1016.383, PB.1016.384) och är inte kopplad till någon av de andra SNP:arna. De två 5′ UTR SNP:erna (chr4: 90757312 och chr4: 90758389) täcks av två ömsesidigt exklusiva exon 1-användningar och är därför inte heller kopplade till varandra.

Tabell 3. Information om SNP:er i cDNA.
Vår nuvarande metod är begränsad till att endast ringa in substitutionsvarianter i transkriberade regioner med tillräcklig täckning. En jämförelse av listan över våra SNP:er med hg19 dbSNP-annotationen visar att de flesta av de SNP:er eller varianter som missades antingen var mindre än 1 % frekventa i populationen, inte var enskilda nukleotidssubstitutioner eller angränsade till regioner med låg komplexitet. Till exempel rapporteras rs77964369 (chr4: 90646532) ha en 50/50-frekvens av T/A, men detta T gränsar till en sträcka på 11 genomiska As nedströms. Manuell inspektion av Iso-Seq-läsningsstapeln, som har ~1 300 läsningar på denna plats, tyder inte på tecken på variation, åtminstone inte bland våra 12 prover.
Med hjälp av de provspecifika läsningarna kallar vi genotypen för varje prov vid varje SNP-plats (tabell 3). Förutom att PD-2 har för få läsningar och är ofullständig för alla fyra SNP:er, kunde vi ringa in genotypen för de flesta andra prover. DLB-3 var det enda prov som var heterozygot på alla SNP-platser. I övrigt observerade vi inget tillståndsspecifikt mönster av att föredra en genotyp framför en annan.
Diskussion
Vi beskriver den första studien som använder riktad anrikning av SNCA-genen på multiplexade gDNA- och cDNA-bibliotek för att studera neurologiska sjukdomar med hjälp av sekvensering med långa läsningar. De långa avläsningslängderna i PacBio Sequel-systemet underlättade sekvenseringen av SNCA-genens fullständiga repertoar av transkriptisoformer. Vi avslöjade mångfalden i användningen av alternativa 5′-startplatser och varierande 3′ UTR-längder och observerade kända exonskippinghändelser, t.ex. exon 3-deletion (SNCA126) och exon 5-deletion (SNCA112). Dessutom identifierades nya alternativa start- och slutplatser inom det stora intron 4 som förutspås bli översatta till nya proteiner. Det är troligt att det höga sekvenseringsdjupet vid riktad fångst, i kombination med förmågan att sekvensera kompletta transkript, gjorde det möjligt för oss att upptäcka dessa tidigare obeskrivna isoformer.
Den biologiska och patologiska betydelsen av de olika SNCA-proteinisoformerna har ännu inte upptäckts helt och hållet. Specifika SNCA-isoformer för posttranslationsmodifiering och splicing har dock förknippats med intracellulära aggregationsbenägenheter (Kalivendi et al., 2010) och uttrycks på olika sätt i mänskliga synukleinopatier (Beyer et al., 2008; Beyer och Ariza, 2012). Studier av SNCA:s posttranslationsmodifiering visade att Lewy-kroppar, det patologiska kännetecknet för synukleinopatier, innehåller rikligt med fosforylerat, nitrerat och monoubiquitinerat SNCA (Kim et al., 2014). Effekterna av posttranskriptionell modifiering på SNCA-aggregation har också studerats. Alternativ splicing föreslogs påverka SNCA-aggregationen. En deletion av antingen exon 3 eller 5 förutsäger funktionella konsekvenser: medan exon 3-deletion (SNCA126) leder till att den N-terminala protein-membraninteraktionsdomänen avbryts, vilket kan leda till mindre aggregering, och exon 5-deletion (SNCA112) kan leda till ökad aggregering på grund av en betydande förkortning av den ostrukturerade C-terminalen (Lee et al., 2001; Beyer, 2006). I den frontala cortexen hos DLB är SNCA112 markant ökad jämfört med kontrollerna (Beyer et al., 2008), medan SNCA126-nivåerna är minskade i den prefrontala cortexen hos DLB-patienter (Beyer et al., 2006). Däremot visade SNCA126-uttrycket ökat i den frontala cortexen i PD-hjärnor och inga signifikanta skillnader i MSA (Beyer et al., 2008). SNCA98 är en hjärnspecifik splicevariant som saknar både exon 3 och 5 och uppvisar olika uttrycksnivåer i olika områden i foster- och vuxenhjärnan. Övexpression av SNCA98 har rapporterats i DLB, PD (Beyer et al., 2007) och MSA (Beyer et al., 2008) frontala cortices jämfört med kontroller. Dessutom har den posttranskriptionella processen som resulterar i alternativ användning av 3′UTR rapporterats ha effekter på mRNA:s stabilitet och lokalisering (Fabian et al., 2010; Rhinn et al., 2012; Yeh och Yong, 2016). Ytterligare undersökningar avseende aggregationsbenägenheten hos de olika kända SNCA-proteinisoformerna och sammansättningen av Lewy-kroppar är motiverade. Vidare lade vår studie grunden för mRNA-kvantifieringsanalyser av de tidigare kända och nya transkriptionerna i ett större urval bestående av försökspersoner med olika klinisk-patologiska stadier med hjälp av flera hjärnregioner från varje försöksperson. Dessa analyser av det hjärnregionsspecifika transkriptomiska landskapet av SNCA i samband med neuropatologisk svårighetsgrad kommer att vara informativa med avseende på den roll som specifika SNCA-transkriptisoformer spelar i utvecklingen av de neuropatologiska stadierna och svårighetsgraden av Lewy-kropparna och Lewy-neuriternas täthet.
I den här uppsatsen har vi fokuserat på att skapa en sekvenserings- och analysstandard för att analysera målinriktade gDNA- och cDNA-data som genererats från samma ämnen. Detta är ett kraftfullt tillvägagångssätt som potentiellt gör det möjligt att fasa gDNA-sekvenserna över hela regionen för en viss gen baserat på heterozygositet i sekvensen för isoformerna av det fullständiga transkriptet i full längd. PacBios riktade gDNA-data i den här studien producerade fasade block som täckte 81 % av 114 kb-regionen centrerad på SNCA, där det längsta fasade blocket översteg 54 kb. Eftersom gDNA-fasning begränsas av avläsningslängd och heterozygositet kommer ökande avläsningslängder sannolikt att generera större fasblock.
gDNA-variantanalys bekräftade kända och identifierade nya korta tandemrepetitioner (STRs) i de introniska regionerna. Tidigare har vi till exempel med hjälp av fasad sekvensering genom kloning och Sanger-sekvensering upptäckt fyra olika haplotyper inom en intronisk CT-rik region som bestod av ett kluster av variabla repetitiva sekvenser (Lutz et al., 2015). Vi visade att en specifik haplotyp, kallad haplotyp 3, gav risk för att utveckla Lewy body-patologi hos Alzheimerpatienter. Här validerade vi sekvensen för denna högpolymorfa region med låg komplexitet och dess fyra definierade haplotyper. Även om vårt urval var litet fanns “haplotyp 3” uteslutande hos sjukdomspatienter (en PD-patient, två DLB-patienter), vilket stämmer överens med våra tidigare resultat. Pilotresultaten och vår tidigare publikation ger förutsättningar för att upprepa associationsanalyserna av synukleinopatier med noggrant definierade, dvs. genom långa läsningar, STRs och strukturella haplotyper med hjälp av en större provstorlek.
Vår artikel visade på förmågan hos PacBio Sequel-systemet att upptäcka nya transkript i full längd och karaktärisera den kompletta repertoaren av transkript i full längd för en gen som är inblandad i en sjukdom. Dessutom visade vi också att gDNA med långa läsningar definierar kortare strukturella varianter och haplotyper, inklusive STRs, mer exakt och att detta kan underlätta upptäckten och valideringen av andra sjukdomsrelaterade varianter än SNPs. Sammantaget är denna nya kunskap mycket värdefull och tillämplig för att främja vår förståelse av de genetiska etiologierna, som kan inbegripa störningar i transkriptlandskapet, som ligger till grund för komplexa mänskliga sjukdomar, inklusive åldersrelaterade neurodegenerativa sjukdomar som synukleinopatier.
Datatillgänglighet
De tre SMRT-cellerna av gDNA-rådata finns tillgängliga på Zenodo.org med doi: 10.5281/zenodo.1560688. Den ena SMRT-cellen av cDNA-rådata finns tillgänglig på Zenodo.org med doi: 10.5281/zenodo.1581809. De bearbetade gDNA- och cDNA-resultaten, inklusive gDNA-varianter och cDNA-isoformer, finns tillgängliga på Zenodo.org med doi: 10.5281/zenodo.3261805.
Författarnas bidrag
OC-F bidrog med utformning och design av studien. ET och WR organiserade sekvensdatabaser, utförde sekvenseringsanalyserna och förberedde alla figurer och tabeller. O-CG och JB hanterade hjärnvävnaderna och förberedelserna av nukleinproverna. TH genererade sekvenseringsdatamängderna. SK utformade och anskaffade reagenserna. OC-F, ET och WR skrev det första utkastet till manuskriptet. OC-F erhöll finansiering. Alla författare bidrog till manuskriptförberedelser, läste och godkände den inlämnade versionen.
Finansiering
Detta arbete finansierades delvis av National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Intressekonfliktutlåtande
ET, WR, TH och SK är eller var anställda av Pacific Biosciences under tiden för studien.
De övriga författarna förklarar att forskningen utfördes i avsaknad av kommersiella eller ekonomiska relationer som skulle kunna tolkas som en potentiell intressekonflikt.
Acknowledgments
Detta manuskript har släppts som ett förtryck på BioRxiv (Tseng et al, 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Supplementary Material
Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona study of aging and neurodegenerative disorders and brain and body donation programme. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Α-synukleinets struktur, posttranslationell modifiering och alternativ splicing som aggregationsförstärkare. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Alpha-synuklein posttranslationell modifiering och alternativ splicing som en utlösande faktor för neurodegeneration. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I. och Ariza, A. (2008). Differentiellt uttryck av alfa-synuklein, parkin och synfilin-1 isoformer vid Lewy body disease. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I. och Ariza, A. (2007). Identifiering och karakterisering av en ny alfa-synuklein-isoform och dess roll i Lewy body-sjukdomar. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Låga alfa-synuklein 126 mRNA-nivåer vid demens med Lewy-kroppar och Alzheimers sjukdom. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N. och Filipowicz, W. (2010). Reglering av mRNA-translation och stabilitet genom mikroRNA. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multipel sekvensanpassning med hög noggrannhet och hög genomströmning. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J. och Kalyanaraman, B. (2010). Oxidanter inducerar alternativ splicing av Α-synuklein: konsekvenser för Parkinsons sjukdom. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., and Halliday, G. M. (2014). Alpha-synukleinbiologi i Lewy body-sjukdomar. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgenreceptorvarianten AR-V9 samuttrycks med AR-V7 i metastaser av prostatacancer och förutsäger abirateronresistens. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). Membranbundet Α-synuklein har en hög aggregeringsbenägenhet och förmågan att sätta igång aggregeringen av den cytosoliska formen. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D. och Chiba-Falek, O. (2015). En cytosin-thymin (CT)-rik haplotyp i intron 4 av SNCA ger risk för Lewy body-patologi vid Alzheimers sjukdom och påverkar SNCA-uttrycket. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnostisering och behandling av demens med Lewykroppar: tredje rapporten från DLB-konsortiet. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Rapport från den andra internationella workshopen om demens med Lewy body: diagnos och behandling. Consortium on dementia with Lewy bodies. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M. och Isacson, O. (2012). Transkriptuttrycksnivåer av alfa-synuklein i full längd och dess tre alternativt splicade varianter i hjärnregioner med Parkinsons sjukdom och i en transgen musmodell för överuttryck av alfa-synuklein. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to ten of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternativ användning av Α-synuklein-transkript som en konvergent mekanism i Parkinsons sjukdomspatologi. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transkriptomisk profilering av renade patientavledda dopaminneuroner identifierar konvergerande störningar och terapier för Parkinsons sjukdom. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R. och Südhof, T. C. (2014). Kartografi av alternativ splicing av neurexin kartlagd med hjälp av single-molecule long-read mRNA-sekvensering. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L. och Tassone, F. (2017). Förändrat uttryck av FMR1-splicingvarianternas landskap hos premutationsbärare. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L. och Yu, J.-T. (2014). Kopplingen mellan SNCA-genen och parkinsonism. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Alternativ polyadenylering av mRNA: 3′-untranslated region har betydelse för genuttryck. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar