- Introduction
- Materialen en Methoden
- Studiemonsters
- Genomisch DNA en RNA-extracties
- Bibliotheek Voorbereiding en Sequencing
- gDNA Capture met behulp van IDT Xgen ® Lockdown ® Probes en Single-Molecule Sequencing
- cDNA Capture met behulp van IDT Xgen ® Lockdown ® Probes en Single-Molecule Isoform-Sequencing (Iso-Seq)
- gDNA Analyse
- Short Variant Analysis and Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Isoform Analysis
- Isoform SNP Calling
- Results
- Targeted gDNA Capture Identified Known and Novel Variations
- Targeted cDNA Capture Identified Novel Start and End Sites
- Full-Length cDNA Enables Isoform-Level Phasing Information
- Discussie
- Data Beschikbaarheid
- Author Contributions
- Funding
- Conflict of Interest Statement
- Acknowledgments
- Aanvullend materiaal
Introduction
Transcriptionele en posttranscriptionele programma’s controleren genexpressieniveaus en/of productie van meerdere verschillende mRNA-isovormen, en veranderingen in deze mechanismen resulteren in ontregeling van genexpressie en differentiële expressieprofielen. Afwijkende transcriptionele en posttranscriptionele genregulatie is overvloedig aanwezig in menselijke zenuwstelselweefsels en draagt bij tot fenotypische verschillen binnen en tussen individuen in gezondheid en ziekte.
Dysregulatie van alpha-synucleine expressie is betrokken bij de pathogenese van synucleinopathieën, in het bijzonder de ziekte van Parkinson (PD) en Dementie met Lewy lichaampjes en (DLB). Terwijl de rol van SNCA overexpressie in synucleinopathieën, voornamelijk PD, goed is aangetoond, hebben wij ons hier gericht op de bepaling van het volledige repertoire van SNCA transcript isovormen in verschillende synucleinopathieën. Eerder zijn verschillende SNCA transcript isovormen beschreven voor SNCA gen, ontstaan door alternatieve splicing, transcriptionele start sites (TSSs), en selectie van polyadenylatie sites (McLean et al., 2012; Xu et al., 2014). Alternatieve splicing van de coderende exonen geeft aanleiding tot SNCA 140, SNCA 112, SNCA 126, en SNCA 98, resulterend in vier eiwit isovormen (Beyer en Ariza, 2012). Alternatieve TSS’s van het SNCA-gen resulteren in vier verschillende 5′UTRs, en alternatieve selectie van verschillende polyadenyleringsplaatsen bepaalt drie grote lengtes van de 3′UTR, zonder invloed op de samenstelling van het eiwitproduct (Beyer en Ariza, 2012). Ons overkoepelende doel is om nieuwe inzichten te verkrijgen in de bijdrage van de verschillende SNCA mRNA soorten, bekende en nieuwe, aan de pathogenese en heterogeniteit van synucleinopathieën.
Tot op heden hebben de meeste studies gebruik gemaakt van short read sequencing technologieën om de transcriptoom complexiteit in menselijke hersenen te ondervragen. De beschikbaarheid van derde generatie long read technologieën geeft een ongekend en bijna compleet beeld van isovorm structuren. Bestaande long read transcript sequencing voor menselijke ziektegenen heeft echter een amplicon-gebaseerde aanpak gebruikt (Treutlein et al., 2014; Kohli, 2017; Tseng et al., 2017). Hoewel deze aanpak succesvol is geweest in het identificeren van complexe alternatieve splicing in menselijke ziektegenen, is het beperkt tot het PCR primerontwerp en zal het geen alternatieve start- en eindsites blootleggen. Gerichte verrijking, zoals door het gebruik van IDT-probes, kan een uitgebreide isovormweergave van interessante genen opleveren tegen lage sequencingkosten. Verder, zeer nauwkeurige full-length transcript leest mogelijk isovorm-specifieke haplotyping.
Hier presenteren we de eerste bekende studie met behulp van gerichte vangst van gDNA en cDNA van de SNCA-gen regio met behulp van PacBio SMRT sequencing. De SNCA-gen regio is ~ 114 kb lang, bestaande uit zes exonen met transcript lengtes rond 3 kb. We hebben 12 menselijke hersenmonsters van PD, DLB en normale controlemonsters gemultiplexed en de gDNA- en cDNA-bibliotheek gesequenced op het PacBio Sequel-systeem. We beschrijven de bio-informatica analyses die gebruikt zijn om SNPs, indels en korte tandem herhalingen te identificeren voor de gDNA capture, en haplotypering op isovormniveau voor de cDNA data. Wij tonen aan dat gerichte capture een kosteneffectieve manier is om gezamenlijk genomische variatie en alternatieve splicing te bestuderen in een ziekte-gerelateerd neuraal gen.
Materialen en Methoden
Studiemonsters
Het studiecohort (N = 12) bestond uit personen met drie door autopsie bevestigde neuropathologische diagnoses: (1) PD (N = 4); (2) DLB (N = 4); en (3) klinisch en neuropathologisch normale proefpersonen (N = 4). Frontale cortex hersenweefsels werden verkregen via de Kathleen Price Bryan Brain Bank (KPBBB) aan Duke University, het Banner Sun Health Research Institute Brain and Body Donation Program (Beach et al., 2015), en het Layton Aging and Alzheimer’s Disease Center aan Oregon Health and Science University. Neuropathologische fenotypes werden bepaald bij postmortaal onderzoek volgens standaard gevestigde methoden volgens de methode en de aanbevelingen voor klinische praktijk van McKeith en collega’s (McKeith et al., 1999, 2005). De dichtheid van de LB pathologie (in een standaard set van hersengebieden) kreeg scores van mild, matig, ernstig, en zeer ernstig. De studiemonsters binnen elke diagnosegroep, PD en DLB, werden zorgvuldig geselecteerd zodat de ernst van de klinisch-pathologische fenotypes binnen elke pathologie vergelijkbaar was. Alle hersenen vertoonden hersenstam-, limbische- en neocorticale Lewy bodies (LBs), terwijl PD ernstige tot zeer ernstige McKeith scores vertoonde in de sub-nigra en de amygdala. Alle hersenen wijzen niet op AD volgens de CERAD criteria en Braak en Braak stadium = II. De neurologisch gezonde hersenmonsters werden verkregen uit postmortale weefsels van klinisch normale proefpersonen die, in de meeste gevallen, binnen een jaar na overlijden werden onderzocht en geen cognitieve stoornis of parkinsonisme bleken te hebben en neuropathologische bevindingen die onvoldoende waren voor het stellen van de diagnose PD, ziekte van Alzheimer (AD), of andere neurodegeneratieve stoornissen. Alle monsters waren blanken. Demografische gegevens en neuropathologie voor deze proefpersonen zijn samengevat in aanvullende tabel 1. Het project werd goedgekeurd door de Duke Institution Review Board (IRB) die een ethische goedkeuring gaf. De methoden werden uitgevoerd in overeenstemming met de relevante richtlijnen en voorschriften.
Genomisch DNA en RNA-extracties
Genomisch DNA werd geëxtraheerd uit hersenweefsels door het standaard Qiagen-protocol (Qiagen, Valencia, CA). Totaal RNA werd geëxtraheerd uit hersenmonsters (100 mg) met TRIzol reagens (Invitrogen, Carlsbad, CA), gevolgd door zuivering met een RNeasy kit (Qiagen, Valencia, CA), volgens het protocol van de fabrikant. gDNA en RNA concentratie werd spectrofotometrisch bepaald, en de kwaliteit van de RNA monsters en het ontbreken van significante degradatie werden bevestigd door metingen van het RNA Integrity Number (RIN, Supplementary Table 1) met behulp van een Agilent Bioanalyzer.
Bibliotheek Voorbereiding en Sequencing
gDNA Capture met behulp van IDT Xgen ® Lockdown ® Probes en Single-Molecule Sequencing
Ongeveer 2 ug van elk gDNA monster werd geschoren tot 6 kb met behulp van de Covaris g-TUBE en geligeerd met gebarcodeerde adapters. Een equimolaire pool van 12-plex gebarcodeerde gDNA bibliotheek (2 pg totaal) werd ingevoerd in de probe-based capture met een op maat ontworpen SNCA gen panel.
Een SMRTBell bibliotheek werd gebouwd met behulp van 626 ng van gevangen en opnieuw gemamplificeerde gDNA1.
cDNA Capture met behulp van IDT Xgen ® Lockdown ® Probes en Single-Molecule Isoform-Sequencing (Iso-Seq)
Over 100-150 ng van totaal RNA per reactie werd reverse transcriptie met behulp van de Clontech SMARTer cDNA synthese kit en 12 monster specifieke gebarcodeerde oligo dT (met PacBio 16mer barcode sequenties, zie Aanvullende Methoden). Drie reverse transcription (RT) reacties werden parallel voor elk monster verwerkt. PCR-optimalisatie werd gebruikt om het optimale amplificatiecyclusaantal voor de downstream grootschalige PCR-reacties te bepalen. Een enkele primer (primer IIA van de Clontech SMARTer kit 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) werd gebruikt voor alle PCR-reacties post-RT. Grootschalige PCR-producten werden afzonderlijk gezuiverd met 1X AMPure PB beads, en de bioanalyzer werd gebruikt voor QC. Een equimolaire pool van 12-plex gebarcodeerde cDNA-bibliotheek (1 ug totaal) werd ingevoerd in de probe-based capture met een op maat ontworpen SNCA-gen panel.
Een SMRTBell-bibliotheek werd gebouwd met behulp van 874 ng van gevangen en opnieuw gemamplificeerde cDNA2. Een SMRT Cell 1M (6 h film) werd gesequenced op de PacBio Sequel platform met behulp van 2.0 chemie.
gDNA Analyse
Sequencing van de barcoded gDNA-gegevens werd uitgevoerd op drie SMRT Cellen 1M met behulp van 2.0 chemie. De gegevens werden gedemultiplexed door het uitvoeren van de Demultiplex Barcodes toepassing in PacBio SMRT Link v6.0.
Short Variant Analysis and Phasing
Circular Consensus Sequence (CCS) leest werden gegenereerd met behulp van SMRT Analysis 6.0 van elke gedemultiplexed dataset en uitgelijnd met het hg19 referentiegenoom met behulp van minimap2. PCR duplicaten van post-capture amplificatie werden geïdentificeerd door mapping eindpunten en gelabeld met behulp van een aangepaste script. Korte varianten werden genoemd met behulp van GATK4 HaplotypeCaller (GATK4 HC) (Poplin et al., 2018). Na een eerste pass van filtering met behulp van dekking diepte en kwaliteit metrics, werden varianten handmatig geïnspecteerd in IGV3. Als varianten niet faseerden met nabijgelegen SNP’s, werden ze handmatig gefilterd. De variantensites die handmatige curatie passeerden, werden gebruikt in combinatie met de gededupliceerde CCS-uitlijningen voor read-backed fasering met WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Subsequenties uitgelijnd op chr4: 90742331-90742559 (hg19) werden voor elk monster geëxtraheerd. Na inspectie van de grootteverdeling van deze subsequenties, werden ze geclusterd op grootte en sequentie gelijkenis met behulp van een combinatie van python en MUSCLE (Edgar, 2004), en een consensus sequentie werd onafhankelijk gegenereerd voor elke cluster.
Custom scripts en workflows verder beschreven in https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Isoform Analysis
Sequencing van de gebarcodeerde cDNA-gegevens was op een SMRT Cell 1M op de PacBio Sequel systeem met behulp van 2.0 chemie. Bio-informatica analyse werd gedaan met behulp van de IsoSeq3 toepassing in de PacBio SMRT Analysis v6.0.0 om hoge kwaliteit, full-length isovorm sequenties te verkrijgen (zie Aanvullende Methoden voor meer informatie).
Isoform SNP Calling
Full-length leest geassocieerd met de uiteindelijke 41 isovormen van alle 12 monsters werden uitgelijnd met het hg19-genoom om een pileup te creëren. Bases met QV minder dan 13 werden uitgesloten. Dan, op elke positie met ten minste 40 base dekking, een Fisher exact test met Bonferroni correctie is toegepast met een p cutoff van 0,01. Alleen substitutie-SNP’s die niet in de buurt van homopolymeerregio’s (stukken van 4 of meer van dezelfde nucleotide) lagen, werden opgeroepen. Na SNP-calling, werd het genotype voor elk monster bepaald door het tellen van het aantal ondersteunende monster-specifieke volledige lengte (FL) leest. Als een monster 5+ FL-lezingen had die zowel de referentie- als de alternatieve basis ondersteunden, was het monster heterozygoot. Als een monster meer dan 5 FL-lezingen had die het ene allel ondersteunde en minder dan 5 FL-lezingen voor het andere, was het homozygoot. Anders was het niet overtuigend. Scripts zijn beschikbaar op: https://github.com/Magdoll/cDNA_Cupcake.
Results
We ontwierpen aangepaste probes voor het SNCA-gen en voerde gerichte vangst van zowel gDNA en cDNA op een multiplexed bibliotheek bestaande uit 12 menselijke hersenen monsters van PD, DLB, en normale controles (figuur 1, supplementaire tabel 1). De gDNA en cDNA bibliotheken werden gesequenced op de PacBio Sequel platform. Bioinformatica-analyse werd uitgevoerd met behulp van PacBio-software, gevolgd door aangepaste analyse.
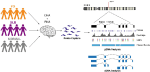
Figuur 1. Schematische weergave van de onderzoeksopzet. Er werd DNA- en RNA-materiaal geëxtraheerd uit postmortaal hersenweefsel van patiënten met de ziekte van Parkinson, dementie met Lewy Body, en controlegroepen. Er werden gDNA- en cDNA-bibliotheken gemaakt met behulp van probe hybridisatie en sequentiebepaling op het PacBio Sequel-systeem. Analyse werd uitgevoerd met behulp van PacBio-software en andere bestaande tools.
Targeted gDNA Capture Identified Known and Novel Variations
Na het genereren van circulaire consensussequenties (CCS) en het verwijderen van PCR-duplicaten (Supplemental Methods), verkregen we een 16- tot 71-voudige gemiddelde unieke dekking van het SNCA-gengebied. De CCS-lezingen hadden een gemiddelde insteeklengte van 2,9 kb en een gemiddelde leesnauwkeurigheid van 98,9%. Met uitzondering van een gebied van 5 kb dat opzettelijk niet door probes werd gedekt vanwege de aanwezigheid van LINE-elementen (hg19 chr4: 90697216-90702113) en een gebied van 2,1 kb met een hoog GC-gehalte rond exon 1, was er voldoende dekking om beide haplotypen voor elk van de 212 monsters te genotyperen (figuur 2, aanvullende figuur 1).

Figuur 2. Gerichte gDNA-opname en fasering. Een voorbeeld met een monster uit elke conditie. Het bovenste spoor toont een van de SNCA-isovormen, gevolgd door de gDNA-dekking voor de drie monsters. De variant spoor toont elke SNP en zijn kleur-gecodeerd voor heterozygote (paars), homozygoot alternatief (oranje), en homozygoot referentie (grijs). Gefaseerde blokken worden in lichtblauw weergegeven. Onderste track toont capture sonde locaties. De dropout regio in probe ontwerp is te wijten aan twee LINE-elementen in het midden van intron 4. Voor de gDNA dekking en fasering informatie van alle 12 monsters, zie aanvullende Figures.
Met behulp van GATK4 HC, kwaliteit-gebaseerde filtering, en handmatige curatie, identificeerden we 282 SNP’s en 35 indels, waaronder 8 SNPS en 13 indels niet gevonden in dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (Supplementary Table 2). Er werden geen varianten geïdentificeerd in de coderende regio voor SNCA, hoewel er acht varianten werden geïdentificeerd in niet-vertaalde regio’s. De meerderheid van de geïdentificeerde varianten, waaronder verschillende korte tandem herhalingen (STR), vallen binnen introns 2, 3, en 4.
We hebben eerder een zeer polymorfe CT-rijke regio in intron 4 van SNCA beschreven met vier waargenomen haplotypen (Lutz et al., 2015). Hoewel deze sterk repetitieve en structureel variabele regio moeilijk te genotyperen bleek met GATK4 HC, waren we in staat om consensussequenties te construeren voor alle 12 monsters en alle 4 van de eerder ontdekte haplotypen waar te nemen (Supplementary Figure 2). Bovendien identificeerden we een nieuwe STR in intron 4 bestaande uit een drie-base-eenheid die 16 keer in de referentie werd herhaald. Binnen de 12 monsters identificeerden we drie haplotypes, met 9, 12, en 15 kopieën van de TTG herhalingseenheid. GATK HC genotypeerde ze allemaal correct, behalve één haplotype voor PD-4, dat een vrij lage dekking had in deze regio. Met de gegeven gegevens voor dit monster kan het genotype echter door visuele inspectie worden bepaald (tabel 1).

Tabel 1. Een nieuwe triplet tandem repeat in intron 4 (chr4: 90713442).
We gebruikten de korte varianten gedetecteerd door GATK HC in combinatie met de lees-gebaseerde faseringstool WhatsHap (Martin et al., 2016) om de CCS-lezen over de locus te faseren, met een reeks van succes vooral gedreven door de heterozygote variantdichtheid over de locus. Monsters PD-1, PD-4, N-4, DLB-1, en DLB-4 hadden lange stukken van lage heterozygositeit, met zeer weinig, korte faseblokken, terwijl de andere monsters faseblokken opleverden variërend van 7 tot 18 keer de gemiddelde leeslengte, tot 54 kb (Supplementary Figure 3).
Targeted cDNA Capture Identified Novel Start and End Sites
We verwerkten de PacBio cDNA (Iso-Seq) -gegevens met behulp van de PacBio SMRT Analysis-software. Na het in kaart brengen van de Iso-Seq gegevens naar hg19 en het verwijderen van artefacten (Supplementary Table 3, Supplementary Figure 4), verkregen we een definitieve set van 41 SNCA isovormen (figuur 3). Alle uiteindelijke isovormen hebben alle canonieke splice sites (GT-AG of GC-AG) en worden ondersteund door een totaal van 20 of meer full-length reads. De meerderheid van de isovormen (28 van de 41) hebben alle zes exonen, en verschillen alleen in het gebruik van alternatieve 5′ start sites en 3′ UTR lengtes. De 3′ UTR lengtes varieerden tussen 300 en 2.6 kb. Het gebruik van zeer uiteenlopende alternatieve 5′ start sites in SNCA is bekend; wat minder bekend is, is de variabele 3′ UTR lengte, die eerder was onderzocht met behulp van RNA-seq gegevens die geen full-length isovorm structuren oplosten (Rhinn et al., 2012). De Iso-Seq data laten zien dat de variabele 3′ UTR lengte gekoppeld lijkt te zijn aan alle mogelijke combinaties van 5′ start sites zonder preferentiële koppeling. Bijna geen van de variabiliteit in start- en eindsite verandert het voorspelde open leesframe (supplementaire figuur 5) en wordt voorspeld te vertalen naar de canonieke 141 aminozuursequentie.

Figuur 3. SNCA-isovormen die met behulp van gerichte Iso-Seq zijn vastgelegd, identificeren nieuwe begin- en eindpunten. Het grootste deel van de isovormcomplexiteit is het gevolg van combinatorisch gebruik van alternatieve 3′ UTR-lengtes en exon 1, met enkele zeldzame alternatieve splitsingsplaatsen in exon 1 (groen), 2 (rood), en 4 (blauw). Alle knooppunten hebben canonieke splitsingsplaatsen. Wij identificeerden vijf isovormen die exon 5 oversloegen en twee isovormen die exon 3 oversloegen. We identificeerden ook nieuwe start- (oranje) en eindpunten (paars) in intron 4. Genoemde SNPs zijn in paars gemarkeerd.
We hebben de nieuwe (maar canonieke) knooppunten verder gevalideerd met behulp van openbaar beschikbare short read knooppuntgegevens. De Intropolis (v1, https://github.com/nellore/intropolis) database combineert meer dan 21.000 openbaar beschikbare RNA-seq. Door het hoge volume van knooppunt gegevens ondersteund door slechts een korte lezen, voor deze studie, moeten we een minimum van 10 korte lezen ondersteuning (gecombineerd uit alle >21,000 RNA-seq datasets) om onze Iso-Seq nieuwe knooppunten te bevestigen. Met uitzondering van de nieuwe knooppunten voor PB.1016.253 en PB.1016.296 (figuur 3), worden alle andere nieuwe knooppunten ondersteund door de Intropolis dataset. Interessant is dat deze nieuwe knooppunten aanzienlijk minder korte gelezen ondersteuning hebben dan de Gencode-geannoteerde knooppunten. Bijvoorbeeld, de twee nieuwe knooppunten in PB.1016.139 geïntroduceerd door het nieuwe exon worden ondersteund door respectievelijk 2.519 en 44 Intropolis short read counts, terwijl de andere vier bekende knooppunten worden ondersteund door meer dan 1 miljoen short read counts. Dit toont de kracht van gerichte verrijking met behulp van full-length transcriptoom sequencing voor het detecteren van zeldzame, nieuwe isovormen.
We observeerden twee isovormen met exon 3 skipping (SNCA126) en vijf isovormen met exon 5 skipping (SNCA112). Ook hier komt de splicing-diversiteit in deze twee exon skipping groepen vooral voort uit het diverse gebruik van alternatieve 5′ start sites en variabele 3′ UTR lengte. ORF-voorspelling toont aan dat het skippen van exon 3 of exon 5 de ORF verkort maar het leesframe handhaaft. Drie isovormen hebben nieuwe 3′ end sites in intron 4. ORF-voorspelling laat zien dat dit resulteert in een afgekapt eiwitproduct.
Wij identificeerden een niet eerder geannoteerde 5′ start site in intron 4 (hg19 chr4: 90692548-90693045, figuur 3). De drie isovormen geassocieerd met deze nieuwe start bestaat uit de nieuwe start site, exon 5, en variabele 3′ UTR lengtes. Interessant is dat, terwijl openbaar gedownloade short read gegevens van GTEx en Sandor et al. (2017) en CAGE piekgegevens (FANTOM5) deze nieuwe startplaats niet ondersteunden, een recente openbare NA12878 directe RNA-dataset4 slechts één SNCA-transcript bevatte dat deze alternatieve startplaats bevestigde. Verder wordt de nieuwe junctie tussen exon 5 en de nieuwe start site bevestigd door Intropolis short read junction gegevens. Interessant is dat deze nieuwe 5′ start site wordt voorspeld om nieuwe peptiden te introduceren met behoud van het leesframe in exon 5.
We identificeerden ook drie SNCA transcripten met nieuwe eindpunten (figuur 3). Twee isovormen (PB.1016.383, PB.1016.384) gebruikten een verlengde 3′ UTR in exon 4, terwijl de derde isovorm (PB.1016.381) een nieuw 3′ exon in intron 4 gebruikte. De nieuwe juncties tussen het nieuwe laatste exon en het vorige exon worden ondersteund door openbare short read junction gegevens (Intropolis). De nieuwe 3′ UTRs resulteren in een afgeknotte ORF voorspelling.
Gebruik makend van de genormaliseerde full-length read count als een proxy voor isovorm abundantie, vinden we een van de canonieke SNCA isovormen (PB.1016.131) als de meest overvloedige, met een abundantie van 50-60% over alle proefpersonen monsters (Supplementary Table 4). We hebben de 41 isovormen verder gegroepeerd naar hun splicingpatronen (Tabel 2). Isovormen die alle zes exonen hebben zijn goed voor 95-97% van de abundantie. Eerdere studies hebben een duidelijke expressieverhoging aangetoond van isovormen die exon 3 missen (SNCA126) in de frontale cortex van DLB-monsters vergeleken met normale monsters (Beyer et al., 2008); onze geaggregeerde isovormtellingen laten zien dat drie van de DLB-monsters een licht verhoogd aantal hebben vergeleken met de normale monsters, evenals de SNCA112 (exon 5 skipping) varianten voor PD en DLB vergeleken met normale monsters.

Tabel 2. SNCA isovorm abundantie voor elk monster, geaggregeerd door splicing patronen.
Full-Length cDNA Enables Isoform-Level Phasing Information
We riepen SNP’s met behulp van cDNA door het stapelen van alle full-length leest van de 12 monsters om varianten te bellen (zie paragraaf “Methoden”). Een totaal van vier SNP’s werden genoemd en alle waren eerder geannoteerd in dbSNP (tabel 3, figuur 3). De vier SNPs bevinden zich allemaal in niet-CDS regio’s, één in de 3′ UTR (exon 6), één in intron 4, en twee in de 5′ UTR (exon 1). De 3′ UTR SNP (chr4: 90646886) wordt alleen gedekt door isovormen met een 3′ UTR dat ten minste ~1 kb lang is, en daarom dekken niet alle canonieke isovormen deze SNP. De intron 4 SNP (chr4: 90743331) wordt alleen gedekt door de nieuwe alternatieve 3′ eind-isovormen (PB.1016.383, PB.1016.384) en is niet verbonden met een van de andere SNPs. De twee 5′ UTR SNPs (chr4: 90757312 en chr4: 90758389) worden gedekt door twee elkaar uitsluitende exon 1 gebruik en zijn dus ook niet gekoppeld.

Tabel 3. cDNA SNP informatie.
Onze huidige aanpak is beperkt tot het oproepen van alleen substitutie varianten in getranscribeerde regio’s met voldoende dekking. Vergelijking van de lijst van onze SNP’s met de hg19 dbSNP annotatie blijkt dat de meeste van de SNP’s of varianten gemist waren ofwel minder dan 1% frequentie in de bevolking, waren niet enkele nucleotide substituties, of grenzend aan lage complexiteit regio’s. Bijvoorbeeld, rs77964369 (chr4: 90646532) wordt gerapporteerd om 50/50 frequentie van T/A te hebben; echter, deze T grenst aan een stuk van 11 genomische As stroomafwaarts. Handmatige inspectie van de Iso-Seq gelezen pileup, die ~ 1.300 leest op deze site heeft, suggereert geen bewijs van variatie althans onder onze 12 monsters.
Met behulp van de steekproef-specifieke leest, noemen we het genotype van elk monster op elke SNP locatie (tabel 3). Behalve bij PD-2, waar te weinig gegevens beschikbaar zijn en het genotype voor alle vier SNP’s onduidelijk is, konden we voor de meeste andere monsters het genotype bepalen. Met name DLB-3 was het enige monster dat heterozygoot is op alle SNP-locaties.
Discussie
Wij beschrijven de eerste studie met gerichte verrijking van het SNCA-gen op multiplexed gDNA- en cDNA-bibliotheken voor het bestuderen van neurologische ziekten met behulp van long read sequencing. De lange leeslengte van het PacBio Sequel systeem vergemakkelijkte de sequenering van het volledige transcript isovormen repertoire van het SNCA gen. We onthulden de diversiteit in het gebruik van alternatieve 5′ start sites en variabele 3′ UTR lengtes en observeerden bekende exon skipping events, zoals exon 3 deletie (SNCA126) en exon 5 deletie (SNCA112). Bovendien werden nieuwe alternatieve start- en eindpunten binnen het grote intron 4 geïdentificeerd, waarvan voorspeld wordt dat ze vertaald worden naar nieuwe eiwitten. Het is waarschijnlijk dat de hoge diepte van sequencing dekking van gerichte capture, in combinatie met de mogelijkheid om volledige transcripten te sequencen, ons in staat stelde om deze voorheen onbeschreven isovormen te detecteren.
De biologische en pathologische betekenis van de verschillende SNCA-eiwit isovormen moet nog volledig worden ontdekt. Echter, specifieke SNCA post translationele modificatie en splicing isovormen zijn geassocieerd met intracellulaire aggregatie neigingen (Kalivendi et al., 2010) en komen verschillend tot expressie in menselijke synucleinopathieën (Beyer et al., 2008; Beyer en Ariza, 2012). Studies van SNCA post translationele modificatie toonden aan dat Lewy bodies, het pathologische kenmerk van synucleinopathieën, overvloedig gefosforyleerd, genitreerd en monoubiquitinated SNCA bevatten (Kim et al., 2014). De effecten van post-transcriptionele modificatie op SNCA aggregatie zijn ook bestudeerd. Alternatieve splicing werd gesuggereerd om SNCA-aggregatie te beïnvloeden. Een deletie van ofwel exon 3 of 5 voorspelt functionele gevolgen: terwijl een exon 3 deletie (SNCA126) leidt tot de onderbreking van het N-terminale eiwit-membraan interactie domein, wat kan leiden tot minder aggregatie, en exon 5 deletie (SNCA112) kan leiden tot verhoogde aggregatie als gevolg van een aanzienlijke verkorting van de ongestructureerde C-terminus (Lee et al., 2001; Beyer, 2006). In de frontale cortex van DLB is SNCA112 duidelijk verhoogd vergeleken met de controles (Beyer et al., 2008), terwijl SNCA126 niveaus verlaagd zijn in de prefrontale cortex van DLB patiënten (Beyer et al., 2006). Daarentegen bleek de expressie van SNCA126 verhoogd in de frontale cortex van PD hersenen en geen significante verschillen in MSA (Beyer et al., 2008). SNCA98 is een hersenspecifieke splice variant die zowel exon 3 als 5 mist en verschillende expressieniveaus vertoont in verschillende gebieden van foetale en volwassen hersenen. Overexpressie van SNCA98 is gerapporteerd in DLB, PD (Beyer et al., 2007), en MSA (Beyer et al., 2008) frontale cortexen vergeleken met controles. Bovendien werd gemeld dat het post-transcriptionele proces dat resulteert in alternatief 3′UTR-gebruik effecten heeft op de mRNA-stabiliteit en -lokalisatie (Fabian et al., 2010; Rhinn et al., 2012; Yeh and Yong, 2016). Verder onderzoek naar de aggregatie neigingen van de verschillende bekende SNCA eiwit isovormen en de samenstelling van Lewy lichaampjes is gerechtvaardigd. Bovendien legde onze studie de basis voor mRNA-kwantificatieanalyses van de eerder bekende en nieuwe transcripten in een grotere steekproefgrootte bestaande uit proefpersonen met een reeks van klinisch-pathologische stadia, waarbij verschillende hersenregio’s van elke proefpersoon werden gebruikt. Deze analyses van het hersenregio-specifieke transcriptomische landschap van SNCA in de context van neuropathologische ernst zullen informatief zijn met betrekking tot de rol van specifieke SNCA transcript isovormen in de progressie van de neuropathologische stadia en de ernst van de Lewy lichaampjes en Lewy neurieten dichtheid.
In dit artikel hebben we ons gericht op het creëren van een sequencing en analyse standaard voor het analyseren van gerichte gDNA en cDNA gegevens gegenereerd van dezelfde proefpersonen. Dit is een krachtige aanpak die mogelijk de fasering van de gDNA-sequenties over de volledige regio van een bepaald gen op basis van heterozygositeit in de sequentie van de full-length transcript isovormen. De PacBio gerichte gDNA gegevens in deze studie geproduceerd gefaseerde blokken die 81% van de 114 kb regio gecentreerd op SNCA gedekt, met de langste gefaseerde blok van meer dan 54 kb. Aangezien gDNA fasering wordt beperkt door leeslengte en heterozygositeit, zullen toenemende leeslengtes waarschijnlijk grotere faseblokken genereren.
gDNA-variantenanalyse bevestigde bekende en identificeerde nieuwe korte tandem herhalingen (STR’s) in de intronic regio’s. Bijvoorbeeld, eerder, met behulp van gefaseerde sequencing door klonering en Sanger-sequencing, ontdekten we vier verschillende haplotypes binnen een intronic CT-rijke regio die bestond uit een cluster van variabele repetitieve sequenties (Lutz et al., 2015). We toonden aan dat een specifiek haplotype, haplotype 3 genoemd, risico gaf op het ontwikkelen van Lewy body pathologie bij Alzheimer patiënten. Hier hebben we de volgorde van deze zeer polymorfe regio met lage complexiteit en de vier gedefinieerde haplotypen gevalideerd. Hoewel onze steekproef klein was, was “haplotype 3” uitsluitend aanwezig bij ziektepatiënten (één PD-patiënt, twee DLB-patiënten), consistent met onze eerdere bevindingen. De pilotresultaten en onze eerdere publicatie bieden de premisse om de associatieanalyses van synucleinopathieën met nauwkeurig gedefinieerde, d.w.z. door long reads, STR’s en structurele haplotypen te herhalen met behulp van een grotere steekproefgrootte.
Onze paper toonde het vermogen aan van het PacBio Sequel-systeem om nieuwe transcripten van volledige lengte te ontdekken en het volledige repertoire van transcripten van volledige lengte van een gen te karakteriseren dat betrokken is bij een ziekte. Bovendien hebben we ook aangetoond dat long reads gDNA korte structurele varianten en haplotypes, waaronder STR’s, nauwkeuriger definiëren en daardoor de ontdekking en validatie van andere ziekte-geassocieerde varianten dan SNP’s kunnen vergemakkelijken. Collectief, deze nieuwe kennis is zeer waardevol en toepasbaar in het bevorderen van ons begrip van de genetische etiologie, die verstoringen in het transcript landschap, onderliggende complexe menselijke ziekten met inbegrip van leeftijdsgebonden neurodegeneratieve aandoeningen zoals synucleinopathies.
Data Beschikbaarheid
De drie SMRT cellen van gDNA ruwe gegevens is beschikbaar op Zenodo.org met doi: 10.5281/zenodo.1560688. De een SMRT cel van cDNA ruwe gegevens is beschikbaar op Zenodo.org met doi: 10.5281/zenodo.1581809. De verwerkte gDNA en cDNA resultaten, met inbegrip van gDNA varianten en cDNA isovormen, zijn beschikbaar op Zenodo.org met doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F bijgedragen concept en ontwerp van de studie. ET en WR georganiseerd sequenties databases, voerde de sequencing analyses en bereidde alle figuren en tabellen. O-CG en JB behandeld de hersenen weefsels en nucleïne monster preparaten. TH gegenereerd de sequencing datasets. SK ontworpen en verkregen van de reagentia. OC-F, ET en WR schreef de eerste versie van het manuscript. OC-F verkregen financiering. Alle auteurs bijgedragen aan manuscript voorbereidingen, gelezen en goedgekeurd de ingediende versie.
Funding
Dit werk werd mede gefinancierd door de National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Conflict of Interest Statement
ET, WR, TH, en SK zijn of waren werknemers van Pacific Biosciences op het moment van het onderzoek.
De overige auteurs verklaren dat het onderzoek werd uitgevoerd in de afwezigheid van enige commerciële of financiële relaties die zouden kunnen worden opgevat als een potentieel belangenconflict.
Acknowledgments
Dit manuscript is vrijgegeven als een pre-print op BioRxiv (Tseng et al., 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Aanvullend materiaal
Het aanvullend materiaal voor dit artikel is online te vinden op: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona studie van veroudering en neurodegeneratieve aandoeningen en hersenen en lichaam donatie programma. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract |Ref Full Text | Google Scholar
Beyer, K. (2006). Α-synucleine structuur, posttranslationele modificatie en alternatieve splicing als aggregatie versterkers. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Alpha-synuclein posttranslationele modificatie en alternatieve splicing als een trigger voor neurodegeneratie. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., and Ariza, A. (2008). Differentiële expressie van alpha-synuclein, parkin, en synphilin-1 isovormen bij de ziekte van Lewy body. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Identificatie en karakterisering van een nieuwe alfa-synucleïne isovorm en zijn rol in Lewy body ziekten. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Lage alfa-synucleïne 126 mRNA niveaus bij dementie met Lewy lichaampjes en de ziekte van Alzheimer. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., and Filipowicz, W. (2010). Regeling van mRNA translatie en stabiliteit door microRNAs. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment met hoge nauwkeurigheid en hoge verwerkingscapaciteit. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Oxidanten induceren alternatieve splicing van Α-synucleïne: implicaties voor de ziekte van Parkinson. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., and Halliday, G. M. (2014). Alpha-synuclein biologie in Lewy body ziekten. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgeenreceptorvariant AR-V9 is coexpressief met AR-V7 in prostaatkankermetastasen en voorspelt abirateronresistentie. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). Membraan-gebonden Α-synucleïne heeft een hoge aggregatie neiging en de mogelijkheid om de aggregatie van de cytosolische vorm te zaaien. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., and Chiba-Falek, O. (2015). Een cytosine-thymine (CT)-rijk haplotype in intron 4 van SNCA confereert risico voor Lewy body pathologie bij de ziekte van Alzheimer en beïnvloedt SNCA expressie. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnostiek en behandeling van dementie met Lewy lichaampjes: derde rapport van het DLB consortium. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Verslag van de tweede internationale workshop over dementie met Lewy-lichaampjes: diagnose en behandeling. Consortium voor dementie met Lewy lichaampjes. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., and Isacson, O. (2012). Transcript expressieniveaus van full-length alfa-synucleïne en zijn drie alternatief gesplicte varianten in hersengebieden van de ziekte van Parkinson en in een transgeen muismodel van alfa-synucleïne overexpressie. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternative Α-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartografie van neurexine alternatieve splicing in kaart gebracht door single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). Altered expression of the FMR1 splicing variants landscape in premutation carriers. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). De link tussen het SNCA-gen en Parkinsonisme. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Alternatieve polyadenylering van mRNAs: 3′-untranslated region matters in gene expression. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar