- Introduction
- Materiały i metody
- Próbki badawcze
- Genomowe DNA i ekstrakcje RNA
- Przygotowanie biblioteki i sekwencjonowanie
- wyłapywanie gDNA przy użyciu IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
- CDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Isoform-Sequencing (Iso-Seq)
- Analiza gDNA
- Short Variant Analysis and Phasing
- Clustering and Determining Haplotypes for CT-Rich Region
- Analiza izoform
- Isoform SNP Calling
- Wyniki
- Targeted gDNA Capture Identified Known and Novel Variations
- Targeted cDNA Capture Identified Novel Start and End Sites
- Full-Length cDNA Enables Isoform-Level Phasing Information
- Dyskusja
- Dostępność danych
- Author Contributions
- Funding
- Oświadczenie o konflikcie interesów
- Podziękowania
- Materiały uzupełniające
Introduction
Transkrypcyjne i posttranskrypcyjne programy kontrolują poziomy ekspresji genów i/lub produkcję wielu odrębnych izoform mRNA, a zmiany w tych mechanizmach powodują dysregulację ekspresji genów i zróżnicowane profile ekspresji. Aberrant transkrypcyjnej i posttranskrypcyjnej regulacji genów jest obfite w tkankach ludzkiego układu nerwowego i przyczynia się do różnic fenotypowych w ramach i między jednostkami w zdrowiu i chorobie.
Dysregulacja ekspresji alfa-synukleiny została implikowana w patogenezie synukleinopatii, w szczególności choroby Parkinsona (PD) i demencji z ciałami Lewy’ego i (DLB). Podczas gdy rola nadekspresji SNCA w synukleinopatiach, głównie PD, została dobrze poznana, tutaj skupiliśmy się na określeniu pełnego repertuaru izoform transkryptu SNCA w różnych synukleinopatiach. Wcześniej opisano kilka różnych izoform transkryptu SNCA dla genu SNCA, powstałych w wyniku alternatywnego splicingu, miejsc startu transkrypcji (TSSs) i selekcji miejsc poliadenylacji (McLean i in., 2012; Xu i in., 2014). Alternatywny splicing eksonów kodujących daje początek SNCA 140, SNCA 112, SNCA 126 i SNCA 98, w wyniku czego powstają cztery izoformy białka (Beyer i Ariza, 2012). Alternatywne TSS genu SNCA skutkuje powstaniem czterech różnych 5′UTR, a alternatywny wybór różnych miejsc poliadenylacji determinuje trzy główne długości 3′UTR, bez wpływu na skład produktu białkowego (Beyer i Ariza, 2012). Naszym nadrzędnym celem jest uzyskanie nowego wglądu w udział różnych gatunków SNCA mRNA, znanych i nowych, w patogenezie i heterogenności synukleinopatii.
Do tej pory większość badań wykorzystywała technologie sekwencjonowania krótkich odczytów do badania złożoności transkryptomu w ludzkich mózgach. Dostępność technologii długich odczytów trzeciej generacji zapewnia bezprecedensowy i prawie kompletny obraz struktur izoform. Jednak istniejące sekwencjonowanie transkryptomu długiego odczytu dla ludzkich genów chorobowych wykorzystuje podejście oparte na amplikonach (Treutlein i in., 2014; Kohli, 2017; Tseng i in., 2017). Chociaż podejście to okazało się skuteczne w identyfikacji złożonego alternatywnego splicingu w genach chorób ludzkich, jest ono ograniczone do projektu primera PCR i nie ujawni alternatywnych miejsc początkowych i końcowych. Ukierunkowane wzbogacanie, takie jak użycie sond IDT, może zapewnić kompleksowy przegląd izoform interesujących genów przy niskich kosztach sekwencjonowania. Ponadto, wysoce dokładne odczyty pełnej długości transkryptu umożliwiają specyficzne dla izoform haplotypowanie.
Przedstawiamy pierwsze znane badanie wykorzystujące ukierunkowane wychwytywanie gDNA i cDNA regionu genu SNCA przy użyciu sekwencjonowania PacBio SMRT. Region genu SNCA ma długość ~114 kb, składa się z sześciu eksonów o długości transkryptu około 3 kb. Wykonaliśmy multipleksowanie 12 próbek ludzkiego mózgu z PD, DLB i normalnych próbek kontrolnych oraz sekwencjonowanie biblioteki gDNA i cDNA w systemie PacBio Sequel. Opisujemy analizy bioinformatyczne zastosowane do identyfikacji SNPs, indeli i krótkich powtórzeń tandemowych dla wychwytu gDNA oraz haplotypowania na poziomie izoform dla danych cDNA. Pokazujemy, że ukierunkowane wychwytywanie jest opłacalnym sposobem wspólnego badania zmienności genomowej i alternatywnego splicingu w związanym z chorobą genie neuronalnym.
Materiały i metody
Próbki badawcze
Kohorta badawcza (N = 12) składała się z osób z trzema potwierdzonymi autopsyjnie diagnozami neuropatologicznymi: (1) PD (N = 4); (2) DLB (N = 4); i (3) klinicznie i neuropatologicznie normalne osoby (N = 4). Tkanki mózgowe kory czołowej uzyskano za pośrednictwem Kathleen Price Bryan Brain Bank (KPBBB) na Uniwersytecie Duke’a, Banner Sun Health Research Institute Brain and Body Donation Program (Beach i in., 2015) oraz Layton Aging and Alzheimer’s Disease Center w Oregon Health and Science University. Fenotypy neuropatologiczne określano w badaniu pośmiertnym według standardowych, dobrze ugruntowanych metod, stosując się do zaleceń metody i praktyki klinicznej McKeitha i współpracowników (McKeith i wsp., 1999, 2005). Gęstość patologii LB (w standardowym zestawie regionów mózgu) była punktowana jako łagodna, umiarkowana, ciężka i bardzo ciężka. Próbki do badań w obrębie każdej grupy diagnostycznej, PD i DLB, zostały starannie dobrane tak, aby nasilenie fenotypów klinopatologicznych było podobne w obrębie każdej patologii. Wszystkie mózgi wykazywały obecność ciał Lewy’ego (LBs) w pniu mózgu, układzie limbicznym i neokorowym, podczas gdy PD wykazywała ciężką lub bardzo ciężką punktację McKeitha w podnamiocie i migdale. Wszystkie mózgi wskazywały na brak AD zgodnie z kryteriami CERAD i stadium Braak i Braak = II. Neurologicznie zdrowe próbki mózgu zostały uzyskane z tkanek pośmiertnych klinicznie normalnych osób, które zostały zbadane, w większości przypadków, w ciągu 1 roku od śmierci i stwierdzono, że nie mają zaburzeń poznawczych lub parkinsonizmu i odkryć neuropatologicznych niewystarczających do rozpoznania PD, choroby Alzheimera (AD) lub innych zaburzeń neurodegeneracyjnych. Wszystkie próbki były białe. Dane demograficzne i neuropatologia dla tych przedmiotów są podsumowane w Tabeli dodatkowej 1. Projekt został zatwierdzony przez Duke Institution Review Board (IRB), który zapewnił zgodę etyczną. Metody zostały przeprowadzone zgodnie z odpowiednimi wytycznymi i przepisami.
Genomowe DNA i ekstrakcje RNA
Genomowe DNA zostało wyekstrahowane z tkanek mózgu przez standardowy protokół Qiagen (Qiagen, Valencia, CA). Całkowite RNA ekstrahowano z próbek mózgu (100 mg) przy użyciu odczynnika TRIzol (Invitrogen, Carlsbad, CA), a następnie oczyszczano zestawem RNeasy (Qiagen, Valencia, CA), zgodnie z protokołem producenta. Stężenie gDNA i RNA określono spektrofotometrycznie, a jakość próbek RNA i brak znaczącej degradacji potwierdzono pomiarami RNA Integrity Number (RIN, Tabela uzupełniająca 1) przy użyciu bioanalizatora Agilent.
Przygotowanie biblioteki i sekwencjonowanie
wyłapywanie gDNA przy użyciu IDT Xgen® Lockdown® Probes and Single-Molecule Sequencing
Około 2 μg każdej próbki gDNA ścinano do 6 kb przy użyciu Covaris g-TUBE i ligowano z adapterami z kodami kreskowymi. Równoważna pula 12-pleksowej biblioteki gDNA z kodem paskowym (łącznie 2 μg) została wprowadzona do wychwytu opartego na sondach z niestandardowym panelem genów SNCA.
Biblioteka SMRTBell została skonstruowana przy użyciu 626 ng wychwyconego i ponownie amplifikowanego gDNA1.
CDNA Capture Using IDT Xgen® Lockdown® Probes and Single-Molecule Isoform-Sequencing (Iso-Seq)
Około 100-150 ng całkowitego RNA na reakcję zostało poddane odwrotnej transkrypcji przy użyciu zestawu do syntezy Clontech SMARTer cDNA i 12 specyficznych dla próbki kodowanych paskowo oligo dT (z 16-merowymi sekwencjami kodów paskowych PacBio, patrz Metody uzupełniające). Trzy reakcje odwrotnej transkrypcji (RT) były przetwarzane równolegle dla każdej próbki. Optymalizacja PCR została wykorzystana do określenia optymalnej liczby cykli amplifikacji dla dalszych reakcji PCR na dużą skalę. Pojedynczy primer (primer IIA z zestawu Clontech SMARTer 5′ AAG CAG TGG TAT CAA CGC AGA GTA C 3′) był używany dla wszystkich reakcji PCR po RT. Produkty PCR o dużej skali były oczyszczane oddzielnie za pomocą kulek 1X AMPure PB, a bioanalizator był używany do kontroli jakości. Równoważna pula 12-pleksowej barcode’owej biblioteki cDNA (łącznie 1 μg) została wprowadzona do wychwytu opartego na sondach z niestandardowym panelem genów SNCA.
Biblioteka SMRTBell została skonstruowana przy użyciu 874 ng wychwyconego i ponownie amplifikowanego cDNA2. Jeden SMRT Cell 1M (film 6 h) został poddany sekwencjonowaniu na platformie PacBio Sequel przy użyciu chemii 2.0.
Analiza gDNA
Sekwencjonowanie danych gDNA z kodem paskowym przeprowadzono na trzech SMRT Cells 1M przy użyciu chemii 2.0. Dane zostały zdemultipleksowane poprzez uruchomienie aplikacji Demultiplex Barcodes w PacBio SMRT Link v6.0.
Short Variant Analysis and Phasing
Circular Consensus Sequence (CCS) reads were generated using SMRT Analysis 6.0 from each demultiplexed data set and aligned to the hg19 reference genome using minimap2. Duplikaty PCR z amplifikacji po przechwyceniu były identyfikowane przez mapowanie punktów końcowych i znakowane przy użyciu niestandardowego skryptu. Krótkie warianty zostały wywołane przy użyciu GATK4 HaplotypeCaller (GATK4 HC) (Poplin i in., 2018). Po pierwszym przejściu filtrowania przy użyciu głębokości pokrycia i metryki jakości, warianty zostały ręcznie sprawdzone w IGV3. Jeśli warianty nie fazowały z pobliskimi SNPs, były ręcznie filtrowane. Miejsca wariantów, które przeszły ręczną kuratelę, zostały użyte w połączeniu z deduplikowanymi wyrównaniami CCS dla read-backed phasing z WhatsHap (Martin et al., 2016).
Clustering and Determining Haplotypes for CT-Rich Region
Subsekwencje wyrównane do chr4: 90742331-90742559 (hg19) zostały wyodrębnione dla każdej próbki. Po sprawdzeniu rozkładu wielkości tych sekwencji, zostały one pogrupowane według wielkości i podobieństwa sekwencji przy użyciu kombinacji Python i MUSCLE (Edgar, 2004), a sekwencja konsensusu została wygenerowana niezależnie dla każdego klastra.
Skrypty niestandardowe i przepływy pracy opisane dalej w https://github.com/williamrowell/Long-reads-Sequencing-of-SNCA-in-Diseases.
Analiza izoform
Sekwencjonowanie danych barcoded cDNA było na jednym SMRT Cell 1M na systemie PacBio Sequel przy użyciu chemii 2.0. Analiza bioinformatyczna została wykonana przy użyciu aplikacji IsoSeq3 w PacBio SMRT Analysis v6.0.0 w celu uzyskania wysokiej jakości, pełnometrażowych sekwencji izoform (więcej informacji w Supplementary Methods).
Isoform SNP Calling
Całodługościowe odczyty związane z końcowymi 41 izoformami ze wszystkich 12 próbek zostały wyrównane do genomu hg19 w celu utworzenia pileupu. Bazy z QV mniejszym niż 13 zostały wykluczone. Następnie, w każdej pozycji o pokryciu co najmniej 40 baz, zastosowano dokładny test Fishera z korektą Bonferroniego, przy p cutoff równym 0,01. Wywoływano tylko SNP substytucyjne, które nie znajdowały się w pobliżu regionów homopolimerowych (odcinki składające się z 4 lub więcej takich samych nukleotydów). Po wywołaniu SNP, genotyp dla każdej próbki został określony poprzez zliczenie liczby wspierających odczytów pełnej długości (FL) specyficznych dla danej próbki. Jeśli próbka miała 5+ odczytów FL wspierających zarówno bazę referencyjną, jak i alternatywną, była heterozygotyczna. Jeśli próbka miała 5+ odczytów FL wspierających jeden allel i mniej niż 5 odczytów FL dla drugiego, była homozygotyczna. W przeciwnym razie, próbka była niejednoznaczna. Skrypty są dostępne pod adresem: https://github.com/Magdoll/cDNA_Cupcake.
Wyniki
Zaprojektowaliśmy niestandardowe sondy do genu SNCA i przeprowadziliśmy ukierunkowane wychwytywanie zarówno gDNA, jak i cDNA na multipleksowanej bibliotece składającej się z 12 próbek ludzkiego mózgu z PD, DLB i normalnych kontroli (Figura 1, Tabela uzupełniająca 1). Biblioteki gDNA i cDNA były sekwencjonowane na platformie PacBio Sequel. Analizę bioinformatyczną przeprowadzono przy użyciu oprogramowania PacBio, a następnie analizy niestandardowej.
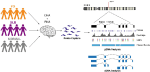
Rysunek 1. Schematyczna prezentacja projektu badania. Materiały DNA i RNA ekstrahowano z pośmiertnych tkanek mózgu pacjentów z chorobą Parkinsona, otępieniem z ciałami Lewy’ego oraz z grup kontrolnych. biblioteki gDNA i cDNA tworzono metodą hybrydyzacji z sondami i sekwencjonowano w systemie PacBio Sequel. Analiza została przeprowadzona przy użyciu oprogramowania PacBio i innych istniejących narzędzi.
Targeted gDNA Capture Identified Known and Novel Variations
Po wygenerowaniu okrągłych sekwencji konsensusu (CCS) i usunięciu duplikatów PCR (Supplemental Methods), uzyskaliśmy 16- do 71-krotne średnie unikalne pokrycie regionu genu SNCA. Odczyty CCS miały średnią długość wstawki 2,9 kb i średnią dokładność odczytu 98,9%. Z wyjątkiem regionu 5 kb celowo odsłoniętego przez sondy z powodu obecności elementów LINE (hg19 chr4: 90697216-90702113) i regionu 2,1 kb o wysokiej zawartości GC wokół eksonu 1, było wystarczające pokrycie do genotypowania obu haplotypów dla każdej z 212 próbek (Figura 2, Dodatkowa Figura 1).

Rysunek 2. Ukierunkowane wychwytywanie i fazowanie gDNA. Przykład pokazujący jedną próbkę z każdego stanu. Górna ścieżka pokazuje jedną z izoform SNCA, a następnie pokrycie gDNA dla trzech próbek. Ścieżka wariantu pokazuje każdy SNP i jest oznaczona kolorem heterozygotycznym (fioletowy), homozygotycznym alternatywnym (pomarańczowy) i homozygotycznym referencyjnym (szary). Bloki fazowe są zaznaczone kolorem jasnoniebieskim. Dolna ścieżka pokazuje lokalizacje sond wychwytujących. Region braku w projekcie sondy jest spowodowany dwoma elementami LINE w środku intronu 4. Dla pokrycia gDNA i informacji o fazowaniu wszystkich 12 próbek, patrz Rysunki uzupełniające.
Używając GATK4 HC, filtrowania opartego na jakości i ręcznej kurateli, zidentyfikowaliśmy 282 SNPs i 35 indeli, w tym 8 SNPS i 13 indeli nie znalezionych w dbSNP (dbSNP Build ID: human_9606_b150_GRCh37p13) (Tabela uzupełniająca 2). Nie zidentyfikowano żadnych wariantów w regionie kodującym dla SNCA, chociaż zidentyfikowano osiem wariantów w regionach nieulegających translacji. Większość zidentyfikowanych wariantów, w tym kilka krótkich powtórzeń tandemowych (STR), mieści się w intronach 2, 3 i 4.
Wcześniej opisaliśmy wysoce polimorficzny region bogaty w CT w intronie 4 SNCA z czterema obserwowanymi haplotypami (Lutz i in., 2015). Chociaż ten wysoce powtarzalny i strukturalnie zmienny region okazał się trudny do genotypowania z GATK4 HC, byliśmy w stanie skonstruować sekwencje konsensusowe dla wszystkich 12 próbek i zaobserwowaliśmy wszystkie 4 z wcześniej odkrytych haplotypów (Supplementary Figure 2). Dodatkowo, zidentyfikowaliśmy nowy STR w intronie 4 składający się z jednostki trójbazowej powtórzonej 16 razy w referencji. W obrębie 12 próbek zidentyfikowaliśmy trzy haplotypy, z 9, 12 i 15 kopiami powtórzonej jednostki TTG. GATK HC poprawnie genotypował wszystkie z nich z wyjątkiem jednego haplotypu dla PD-4, który miał dość niskie pokrycie w tym regionie. Jednakże, przy podanych danych dla tej próbki, genotyp może być określony przez oględziny (Tabela 1).

Tabela 1. A novel triplet tandem repeat in intron 4 (chr4: 90713442).
Użyliśmy krótkich wariantów wykrytych przez GATK HC w połączeniu z narzędziem do fazowania opartym na odczycie WhatsHap (Martin i in., 2016) do fazowania odczytów CCS w całym locus, z zakresem sukcesu napędzanym głównie przez gęstość wariantów heterozygotycznych w locus. Próbki PD-1, PD-4, N-4, DLB-1 i DLB-4 miały długie odcinki o niskiej heterozygotyczności, z bardzo nielicznymi, krótkimi blokami fazowymi, podczas gdy pozostałe próbki dawały bloki fazowe o długości od 7 do 18 razy większej od średniej długości odczytu, aż do 54 kb (Dodatkowa Figura 3).
Targeted cDNA Capture Identified Novel Start and End Sites
Przetworzyliśmy dane PacBio cDNA (Iso-Seq) przy użyciu oprogramowania PacBio SMRT Analysis. Po mapowaniu danych Iso-Seq do hg19 i usunięciu artefaktów (Tabela uzupełniająca 3, Rysunek uzupełniający 4), uzyskaliśmy ostateczny zestaw 41 izoform SNCA (Figura 3). Wszystkie końcowe izoformy mają wszystkie kanoniczne miejsca splice (GT-AG lub GC-AG) i są wspierane przez łącznie 20 lub więcej odczytów o pełnej długości. Większość izoform (28 z 41) posiada wszystkie sześć eksonów, różniąc się jedynie wykorzystaniem alternatywnych miejsc startowych 5′ i długością 3′ UTR. Długości 3′ UTR wahały się między 300 a 2,6 kb. Wykorzystanie bardzo zróżnicowanych alternatywnych miejsc 5′ startu w SNCA jest znane; mniej znana jest natomiast zmienna długość 3′ UTR, która była wcześniej badana przy użyciu danych RNA-seq, które nie pozwoliły na określenie struktury izoform o pełnej długości (Rhinn i in., 2012). Dane Iso-Seq pokazują, że zmienna długość 3′ UTR wydaje się być sparowana ze wszystkimi możliwymi kombinacjami miejsc startu 5′, bez preferencyjnego sprzężenia. Prawie żadna ze zmienności w miejscu startu i końca nie zmienia przewidywanej otwartej ramki odczytu (Supplementary Figure 5) i przewiduje się, że przekłada się na kanoniczną sekwencję 141 aminokwasów.

Figure 3. Izoformy SNCA wychwycone przy użyciu ukierunkowanej Iso-Seq identyfikują nowe miejsca początkowe i końcowe. Większość złożoności izoform pochodzi z kombinatorycznego wykorzystania alternatywnych długości 3′ UTR i eksonu 1, z kilkoma rzadkimi alternatywnymi miejscami splice’owania znalezionymi w eksonie 1 (zielony), 2 (czerwony) i 4 (niebieski). Wszystkie złącza mają kanoniczne miejsca splice. Zidentyfikowaliśmy pięć izoform, które pominęły ekson 5 i dwie izoformy, które pominęły ekson 3. Zidentyfikowaliśmy również nowe miejsca początkowe (pomarańczowe) i końcowe (fioletowe) w intronie 4. Wywołane SNPs są zaznaczone na fioletowo.
Dalej walidowaliśmy nowe (ale kanoniczne) węzły przy użyciu publicznie dostępnych danych z krótkich odczytów węzłów. Baza danych Intropolis (v1, https://github.com/nellore/intropolis) łączy ponad 21 000 publicznie dostępnych danych RNA-seq. Ze względu na dużą ilość danych połączeń wspieranych tylko przez pojedynczy krótki odczyt, do tego badania wymagamy minimum 10 krótkich odczytów (połączonych ze wszystkich >21 000 zbiorów danych RNA-seq), aby potwierdzić nasze nowe połączenia Iso-Seq. Z wyjątkiem nowych połączeń dla PB.1016.253 i PB.1016.296 (Rysunek 3), wszystkie inne nowe połączenia są wspierane przez zestaw danych Intropolis. Co ciekawe, te nowe połączenia mają znacznie mniejsze wsparcie w krótkich odczytach niż połączenia anotowane przez Gencode. Na przykład, dwa nowe połączenia w PB.1016.139 wprowadzone przez nowy ekson są wspierane odpowiednio przez 2519 i 44 krótkie odczyty Intropolis, podczas gdy pozostałe cztery znane połączenia są wspierane przez ponad 1 milion krótkich odczytów. Pokazuje to moc ukierunkowanego wzbogacania przy użyciu sekwencjonowania transkryptomu na całej długości do wykrywania rzadkich, nowych izoform.
Zaobserwowaliśmy dwie izoformy z pominięciem eksonu 3 (SNCA126) i pięć izoform z pominięciem eksonu 5 (SNCA112). Ponownie, różnorodność splicingowa w tych dwóch grupach z pominięciem eksonu pochodzi głównie z różnorodnego wykorzystania alternatywnych miejsc startowych 5′ i zmiennej długości 3′ UTR. Przewidywanie ORF wskazuje, że pominięcie eksonu 3 lub eksonu 5 skraca ORF, ale zachowuje ramkę odczytu. Trzy izoformy mają nowe miejsca 3′ końca zlokalizowane w intronie 4. Przewidywanie ORF pokazuje, że skutkuje to obciętym produktem białkowym.
Zidentyfikowaliśmy wcześniej nienotowane 5′ miejsce startowe zlokalizowane w intronie 4 (hg19 chr4: 90692548-90693045, Figura 3). Trzy izoformy związane z tym nowym startem składają się z nowego miejsca startu, eksonu 5 i zmiennych długości 3′ UTR. Co ciekawe, podczas gdy publicznie pobrane krótkie dane odczytu z GTEx i Sandor et al. (2017) oraz dane szczytowe CAGE (FANTOM5) nie wspierały tego nowego miejsca startu, niedawny publiczny zestaw danych NA12878 direct RNA4 zawierał tylko jeden transkrypt SNCA, który potwierdził to alternatywne miejsce startu. Ponadto, nowe połączenie między eksonem 5 i nowym miejscem startu jest potwierdzone przez dane Intropolis short read junction. Co ciekawe, przewiduje się, że to nowe miejsce startu 5′ wprowadza nowe peptydy przy zachowaniu ramki odczytu w eksonie 5.
Zidentyfikowaliśmy również trzy transkrypty SNCA z nowymi miejscami końcowymi (Figura 3). Dwie izoformy (PB.1016.383, PB.1016.384) używały przedłużonego 3′ UTR w eksonie 4, podczas gdy trzecia izoforma (PB.1016.381) używała nowego 3′ eksonu w intronie 4. Nowe połączenia między nowym ostatnim eksonem a poprzednim eksonem są wspierane przez publiczne dane o połączeniach krótkich odczytów (Intropolis). Nowe 3′ UTR skutkują okrojonym przewidywaniem ORF.
Używając znormalizowanej liczby pełnometrażowych odczytów jako przybliżenia dla obfitości izoform, znajdujemy jedną z kanonicznych izoform SNCA (PB.1016.131), aby być najbardziej obfitą, z obfitością 50-60% we wszystkich badanych próbkach (Tabela uzupełniająca 4). Dalej pogrupowaliśmy 41 izoform według ich wzorów splicingu (Tabela 2). Izoformy, które mają wszystkie sześć eksonów, stanowią 95-97% liczebności. Poprzednie badania wykazały wyraźny wzrost ekspresji izoform pozbawionych eksonu 3 (SNCA126) w korze czołowej próbek DLB w porównaniu do normalnych (Beyer i in., 2008); nasze zagregowane liczenie izoform pokazuje, że trzy z próbek DLB mają nieznacznie podwyższony poziom liczenia w porównaniu do normalnych próbek, jak również warianty SNCA112 (pomijanie eksonu 5) dla PD i DLB w porównaniu do normalnych próbek.

Tabela 2. Obfitość izoform SNCA dla każdej próbki, zagregowana według wzorców splicingu.
Full-Length cDNA Enables Isoform-Level Phasing Information
Wywołaliśmy SNPs używając cDNA poprzez spiętrzenie wszystkich odczytów o pełnej długości z 12 próbek w celu wywołania wariantów (patrz Sekcja “Metody”). W sumie cztery SNP zostały wywołane i wszystkie były wcześniej anotowane w dbSNP (Tabela 3, Rysunek 3). Wszystkie cztery SNP są zlokalizowane w regionach nie-CDS, jeden w 3′ UTR (ekson 6), jeden w intronie 4 i dwa w 5′ UTR (ekson 1). SNP w 3′ UTR (chr4: 90646886) jest pokrywany tylko przez izoformy, których 3′ UTR ma długość co najmniej ~1 kb, a zatem nie wszystkie kanoniczne izoformy pokrywają ten SNP. SNP intronu 4 (chr4: 90743331) jest pokrywany tylko przez nowe alternatywne izoformy 3′ końca (PB.1016.383, PB.1016.384) i nie jest związany z żadnym z pozostałych SNP. Dwa 5′ UTR SNPs (chr4: 90757312 i chr4: 90758389) są objęte przez dwa wzajemnie wykluczające się użycia eksonu 1 i stąd również nie są powiązane.

Tabela 3. Informacje o SNP cDNA.
Nasze obecne podejście jest ograniczone do wywoływania tylko wariantów substytucji w regionach transkrypcyjnych o wystarczającym pokryciu. Porównanie listy naszych SNP z anotacją hg19 dbSNP pokazuje, że większość pominiętych SNP lub wariantów miała częstotliwość mniejszą niż 1% w populacji, nie była pojedynczą substytucją nukleotydową lub przylegała do regionów o niskiej złożoności. Na przykład, rs77964369 (chr4: 90646532) ma częstotliwość 50/50 T/A; jednak to T sąsiaduje z odcinkiem 11 genomów As downstream. Ręczna inspekcja stosu odczytów Iso-Seq, który ma ~ 1300 odczytów w tym miejscu, nie sugeruje dowodów na zmienność przynajmniej wśród naszych 12 próbek.
Używając odczytów specyficznych dla próbki, nazywamy genotyp każdej próbki w każdej lokalizacji SNP (Tabela 3). Poza PD-2, który ma zbyt mało odczytów i jest niejednoznaczny dla wszystkich czterech SNP, byliśmy w stanie wywołać genotyp dla większości innych próbek. Warto zauważyć, że DLB-3 była jedyną próbką, która jest heterozygotyczna we wszystkich lokalizacjach SNP. Poza tym nie zaobserwowaliśmy żadnego specyficznego dla danego stanu wzorca preferowania jednego genotypu względem drugiego.
Dyskusja
Opisujemy pierwsze badanie wykorzystujące ukierunkowane wzbogacanie genu SNCA na multipleksowanych bibliotekach gDNA i cDNA do badania chorób neurologicznych przy użyciu sekwencjonowania długich odczytów. Długie odczyty systemu PacBio Sequel ułatwiły sekwencjonowanie repertuaru izoform transkryptu genu SNCA o pełnej długości. Ujawniliśmy różnorodność w stosowaniu alternatywnych miejsc startu 5′ i zmiennych długości 3′ UTR oraz zaobserwowaliśmy znane przypadki pomijania eksonów, takie jak delecja eksonu 3 (SNCA126) i delecja eksonu 5 (SNCA112). Dodatkowo, zidentyfikowano nowe alternatywne miejsca początku i końca w obrębie dużego intronu 4, które mogą być tłumaczone na nowe białka. Jest prawdopodobne, że duża głębokość pokrycia sekwencjonowania ukierunkowanego wychwytywania, w połączeniu ze zdolnością do sekwencjonowania kompletnych transkryptów, pozwoliła nam wykryć te wcześniej nieopisane izoformy.
Biologiczne i patologiczne znaczenie różnych izoform białka SNCA nie zostało jeszcze w pełni odkryte. Jednakże specyficzne izoformy SNCA ulegające modyfikacji po translacji i splicingowi zostały powiązane z wewnątrzkomórkowymi skłonnościami do agregacji (Kalivendi i in., 2010) i są różnie wyrażone w ludzkich synukleinopatiach (Beyer i in., 2008; Beyer i Ariza, 2012). Badania modyfikacji posttranslacyjnej SNCA wykazały, że ciała Lewy’ego, patologiczny wyróżnik synukleinopatii, zawierają obfite fosforylowane, nitrowane i monoubikwitynowane SNCA (Kim i in., 2014). Badano również wpływ modyfikacji posttranskrypcyjnych na agregację SNCA. Zasugerowano, że alternatywny splicing może wpływać na agregację SNCA. Delecja eksonu 3 lub 5 przewiduje konsekwencje funkcjonalne: podczas gdy delecja eksonu 3 (SNCA126) prowadzi do przerwania N-końcowej domeny interakcji białko-membrana, co może prowadzić do mniejszej agregacji, a delecja eksonu 5 (SNCA112) może skutkować zwiększoną agregacją ze względu na znaczne skrócenie niestrukturalnego C-końca (Lee i in., 2001; Beyer, 2006). W korze czołowej chorych na DLB poziom SNCA112 jest znacznie podwyższony w porównaniu z grupą kontrolną (Beyer i in., 2008), podczas gdy poziom SNCA126 jest obniżony w korze przedczołowej chorych na DLB (Beyer i in., 2006). Z kolei ekspresja SNCA126 wykazała wzrost w korze czołowej mózgów chorych na PD i brak istotnych różnic w MSA (Beyer i in., 2008). SNCA98 jest specyficznym dla mózgu wariantem splice, w którym brakuje eksonu 3 i 5 i wykazuje różny poziom ekspresji w różnych obszarach płodowego i dorosłego mózgu. W DLB, PD (Beyer i in., 2007) i MSA (Beyer i in., 2008) stwierdzono nadekspresję SNCA98 w korze czołowej w porównaniu z grupą kontrolną. Ponadto wykazano, że proces posttranskrypcyjny skutkujący alternatywnym wykorzystaniem 3′UTR ma wpływ na stabilność i lokalizację mRNA (Fabian i in., 2010; Rhinn i in., 2012; Yeh i Yong, 2016). Uzasadnione są dalsze badania dotyczące skłonności do agregacji różnych znanych izoform białka SNCA i składu ciał Lewy’ego. Ponadto, nasze badanie stworzyło podstawy do analizy kwantyfikacji mRNA znanych i nowych transkryptów w większej próbie składającej się z osób o różnym stopniu zaawansowania klinopatologicznego, z wykorzystaniem kilku regionów mózgu od każdego uczestnika. Te analizy specyficznego dla regionu mózgu transkryptomicznego krajobrazu SNCA w kontekście ciężkości neuropatologicznej będą pouczające w odniesieniu do roli specyficznych izoform transkryptu SNCA w progresji etapów neuropatologicznych i ciężkości ciał Lewy’ego i gęstości neurytów Lewy’ego.
W tej pracy skupiliśmy się na tworzeniu standardu sekwencjonowania i analizy dla analizy ukierunkowanych danych gDNA i cDNA wygenerowanych od tych samych podmiotów. Jest to potężne podejście, które potencjalnie umożliwia fazowanie sekwencji gDNA w całym regionie danego genu w oparciu o heterozygotyczność w sekwencji izoform transkryptu o pełnej długości. Dane PacBio targeted gDNA w tym badaniu dały bloki fazowane, które pokryły 81% regionu 114 kb skupionego na SNCA, z najdłuższym blokiem fazowanym przekraczającym 54 kb. Ponieważ fazowanie gDNA jest ograniczone przez długość odczytu i heterozygotyczność, zwiększenie długości odczytu prawdopodobnie wygeneruje większe bloki fazowe.
Analiza wariantów gDNA potwierdziła znane i zidentyfikowała nowe krótkie powtórzenia tandemowe (STRs) w regionach intronowych. Na przykład, poprzednio, używając fazowego sekwencjonowania przez klonowanie i sekwencjonowanie Sangera, odkryliśmy cztery odrębne haplotypy w obrębie intronowego regionu bogatego w CT, który składał się z klastra zmiennych sekwencji powtarzalnych (Lutz i in., 2015). Wykazaliśmy, że specyficzny haplotyp, nazwany haplotypem 3, wiąże się z ryzykiem rozwoju patologii ciał Lewy’ego u pacjentów z chorobą Alzheimera. Tutaj zwalidowaliśmy sekwencję tego wysoce polimorficznego regionu o niskiej złożoności i jego cztery zdefiniowane haplotypy. Chociaż nasza próba była mała, “haplotyp 3” był obecny wyłącznie u pacjentów z chorobą (jeden pacjent z PD, dwóch pacjentów z DLB), zgodnie z naszymi wcześniejszymi odkryciami. Wyniki pilotażowe i nasza poprzednia publikacja stanowią przesłankę do powtórzenia analiz asocjacyjnych synukleinopatii z dokładnie określonymi, tj. przez długie odczyty, STR i haplotypami strukturalnymi przy użyciu większej wielkości próby.
Nasza praca wykazała zdolność systemu PacBio Sequel do odkrywania nowych transkryptów o pełnej długości i charakteryzowania pełnego repertuaru transkryptów o pełnej długości genu implikowanego przez chorobę. Co więcej, wykazaliśmy również, że długie odczyty gDNA dokładniej definiują krótkie warianty strukturalne i haplotypy, w tym STRs, co może ułatwić odkrycie i walidację wariantów związanych z chorobą, innych niż SNP. Łącznie, ta nowa wiedza jest bardzo cenna i ma zastosowanie w zwiększaniu naszego zrozumienia etiologii genetycznych, które mogą obejmować zaburzenia w krajobrazie transkryptów, leżących u podstaw złożonych chorób człowieka, w tym zaburzeń neurodegeneracyjnych związanych z wiekiem, takich jak synukleinopatie.
Dostępność danych
Trzy komórki SMRT surowych danych gDNA są dostępne na Zenodo.org z doi: 10.5281/zenodo.1560688. Jedna komórka SMRT surowych danych cDNA jest dostępna na Zenodo.org z doi: 10.5281/zenodo.1581809. The processed gDNA and cDNA results, including gDNA variants and cDNA isoforms, are available at Zenodo.org with doi: 10.5281/zenodo.3261805.
Author Contributions
OC-F contributed conception and design of the study. ET i WR zorganizowali bazy danych sekwencji, przeprowadzili analizy sekwencjonowania i przygotowali wszystkie ryciny i tabele. O-CG i JB zajęli się przygotowaniem tkanek mózgu i próbek nukleinowych. TH wygenerował zestawy danych sekwencjonowania. SK zaprojektował i uzyskał odczynniki. OC-F, ET i WR napisali pierwszy szkic manuskryptu. OC-F uzyskał finansowanie. Wszyscy autorzy przyczynili się do przygotowania manuskryptu, przeczytali i zatwierdzili przedłożoną wersję.
Funding
Ta praca została częściowo sfinansowana przez National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) .
Oświadczenie o konflikcie interesów
ET, WR, TH i SK są lub byli pracownikami Pacific Biosciences w czasie przeprowadzania badania.
Pozostali autorzy deklarują, że badania zostały przeprowadzone przy braku jakichkolwiek komercyjnych lub finansowych relacji, które mogłyby być interpretowane jako potencjalny konflikt interesów.
Podziękowania
Ten manuskrypt został wydany jako pre-print w BioRxiv (Tseng i in., 2019). https://www.biorxiv.org/content/10.1101/524827v1.
Materiały uzupełniające
Materiały uzupełniające do tego artykułu można znaleźć online pod adresem: https://www.frontiersin.org/articles/10.3389/fgene.2019.00584/full#supplementary-material
Beach, T. G., Adler, C. H., Sue, L. I., Serrano, G., Shill, H. A., Walker, D. G., et al. (2015). Arizona badanie starzenia się i zaburzeń neurodegeneracyjnych oraz program dawstwa mózgu i ciała. Neuropathology 35, 354-389. doi: 10.1111/neup.12189
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K. (2006). Struktura Α-synukleiny, modyfikacje potranslacyjne i alternatywne splicing jako czynniki zwiększające agregację. Acta Neuropathol. 112, 237-251. doi: 10.1007/s00401-006-0104-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., and Ariza, A. (2012). Alpha-synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol. Neurobiol. 47, 509-524. doi: 10.1007/s12035-012-8330-5
CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Humbert, J., Carrato, C., Ferrer, I., and Ariza, A. (2008). Differential expression of alpha-synuclein, parkin, and synphilin-1 isoforms in Lewy body disease. Neurogenetics 9, 163-172. doi: 10.1007/s10048-008-0124-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Beyer, K., Domingo-Sàbat, M., Lao, J. I., Carrato, C., Ferrer, I., and Ariza, A. (2007). Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics 9, 15-23. doi: 10.1007/s10048-007-0106-0
CrossRef Full Text | Google Scholar
Beyer, K., Humbert, J., Ferrer, A., Lao, J. I., Carrato, C., pez, D. L., et al. (2006). Low alpha-synuclein 126 mRNA levels in dementia with Lewy bodies and Alzheimer disease. Neuroreport 17, 1327-1330. doi: 10.1097/01.wnr.0000224773.66904.e7
PubMed Abstract | CrossRef Full Text | Google Scholar
Fabian, M. R., Sonenberg, N., and Filipowicz, W. (2010). Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 79, 351-379. doi: 10.1146/annurev-biochem-060308-103103
PubMed Abstract | CrossRef Full Text | Google Scholar
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792-1797. doi: 10.1093/nar/gkh340
PubMed Abstract | CrossRef Full Text | Google Scholar
Kalivendi, S. V., Yedlapudi, D., Hillard, C. J., and Kalyanaraman, B. (2010). Oxidants induce alternative splicing of Α-synuclein: implications for Parkinson’s disease. Free Radic. Biol. Med. 48, 377-383. doi: 10.1016/j.freeradbiomed.2009.10.045
PubMed Abstract | CrossRef Full Text | Google Scholar
Kim, W. S., Gedal, K. K., and Halliday, G. M. (2014). Biologia alfa-synukleiny w chorobach ciał Lewy’ego. Alzheimers Res. Ther. 6, 1-9. doi: 10.1186/s13195-014-0073-2
CrossRef Full Text | Google Scholar
Kohli, M. (2017). Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin. Cancer Res. 23, 1-13. doi: 10.1158/1078-0432.CCR-17-0017
CrossRef Full Text | Google Scholar
Lee, H.-J., Choi, C., and Lee, S. J. (2001). Związana z błoną Α-synukleina ma wysoką skłonność do agregacji i zdolność do rozsiewania agregacji formy cytozolowej. J. Biol. Chem. 277, 671-678. doi: 10.1074/jbc.M107045200
CrossRef Full Text | Google Scholar
Lutz, M. W., Saul, R., Linnertz, C., Glenn, O.-C., Roses, A. D., and Chiba-Falek, O. (2015). A cytosine-thymine (CT)-rich haplotype in intron 4 of SNCA confers risk for Lewy body pathology in Alzheimer’s disease and affects SNCA expression. Alzheimers Dement. 11, 1133-1143. doi: 10.1016/j.jalz.2015.05.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Martin, M., Patterson, M., Garg, S., Fischer, S. O., Pisanti, N., Klau, G. W., et al. (2016). WhatsHap: fast and accurate read-based phasing. bioRxiv . doi: 10.1101/085050
CrossRef Full Text | Google Scholar
McKeith, I. G., Dickson, D. W., Lowe, J., Emre, M., O’Brien, J. T., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 65, 1863-1872. doi: 10.1212/01.wnl.0000187889.17253.b1
PubMed Abstract | CrossRef Full Text | Google Scholar
McKeith, I. G., Perry, E. K., and Perry, R. H. (1999). Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on dementia with Lewy bodies. Neurology 53, 902-905. doi: 10.1212/WNL.53.5.902
PubMed Abstract | CrossRef Full Text | Google Scholar
McLean, J. R., Hallett, P. J., Cooper, O., Stanley, M., and Isacson, O. (2012). Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol. Cell. Neurosci. 49, 230-239. doi: 10.1016/j.mcn.2011.11.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Poplin, R., Ruano-Rubio, V., DePristo, M. A., Fennell, T. J., Carneiro, M. O., Van der Auwera, G. A., et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv . doi: 10.1101/201178
CrossRef Full Text | Google Scholar
Rhinn, H., Qiang, L., Yamashita, T., Rhee, D., Zolin, A., Vanti, W., et al. (2012). Alternative Α-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 3, 889-821. doi: 10.1038/ncomms2032
CrossRef Full Text | Google Scholar
Sandor, C., Robertson, P., Lang, C., Heger, A., Booth, H., Vowles, J., et al. (2017). Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum. Mol. Genet. 54, ddw412-ddw415. doi: 10.1093/hmg/ddw412
CrossRef Full Text | Google Scholar
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. 111, E1291-E1299. doi: 10.1073/pnas.1403244111
PubMed Abstract | CrossRef Full Text | Google Scholar
Tseng, E., Rowell, W. J., Omolara-Chinue, G., Hon, T., Barrera, J., Kujawa, S., et al. (2019). The landscape of SNCA transcripts across synucleinopathies: new insights from long reads sequencing analysis. bioRxiv . doi: 10.1101/524827
CrossRef Full Text | Google Scholar
Tseng, E., Tang, H.-T., AlOlaby, R. R., Hickey, L., and Tassone, F. (2017). Altered expression of the FMR1 splicing variants landscape in premutation carriers. Biochim. Biophys. Acta Gene. Regul. Mech. 1860, 1117-1126. doi: 10.1016/j.bbagrm.2017.08.007
CrossRef Full Text | Google Scholar
Xu, W., Tan, L., and Yu, J.-T. (2014). Związek między genem SNCA a parkinsonizmem. Neurobiol. Aging 36, 1-14. doi: 10.1016/j.neurobiolaging.2014.10.042
CrossRef Full Text | Google Scholar
Yeh, H.-S., and Yong, J. (2016). Alternatywna poliadenylacja mRNA: 3′-untranslated region matters in gene expression. Mol. Cell 39, 281-285. doi: 10.14348/molcells.2016.0035
PubMed Abstract | CrossRef Full Text | Google Scholar
.