ChIP-seq jest wspaniałą techniką, która pozwala nam badać fizyczne interakcje wiązania między białkiem a DNA przy użyciu sekwencjonowania następnej generacji. W tym artykule dokonam krótkiego przeglądu ChIP i przedstawię technikę sekwencjonowania immunoprecypitacji chromatyny (ChIP-seq), która łączy ChIP z sekwencjonowaniem następnej generacji.
Co to jest immunoprecypitacja chromatyny?
Imprecypitacja chromatyny (ChIP) pozwala nam określić miejsca wiązania białek na DNA. Chromatyna to kompleks DNA upakowany z białkami histonowymi w nukleosomy. ChIP wykorzystuje odwracalne wiązania krzyżowe powstałe pomiędzy DNA i związanymi z nim białkami w wyniku utrwalania komórek lub tkanek formaldehydem. Utrwalona chromatyna jest fizycznie przecinana, a fragmenty DNA związane z określonym białkiem są selektywnie immunoprecypitowane i analizowane. Analiza może być przeprowadzana locus-by-locus przy użyciu PCR, ale częściej ChIP jest badany przy użyciu mikromacierzy (ChIP-chip) lub sekwencjonowania następnej generacji (ChIP-seq).
Jak działa ChIP-chip?
ChIP-on-chip, lub ChIP-chip, łączy immunoprecypitację chromatyny z analizą mikromacierzy (chip). W 2001 roku Jason Lieb z laboratorium Pata Browna w Stanford opublikował pierwszą pracę na temat ChIP-chip (Lieb 2001). W tej metodzie, fragmenty DNA, które wytrącają się z danym białkiem są nanoszone na chip mikromacierzy w celu analizy. W ten sposób uzyskuje się globalny obraz tego, gdzie białko się wiąże, w przeciwieństwie do prostego badania pojedynczych miejsc wiązania za pomocą PCR. Chociaż było to rewolucyjne podejście, technika ta jest ograniczona przez zastosowane technologie macierzy. Po pierwsze, potrzebna jest mikromacierz dla genomu, który chcemy badać, a na początku i w połowie XXI wieku często nie było to możliwe. Po drugie, mikromacierze te są ograniczone przez jakość genomu referencyjnego i zdolność do projektowania sond, które będą działać na tablicy. Po trzecie, tablice są ograniczone pod względem tego, jak duża część genomu może być reprezentowana, i zazwyczaj są one kafelkowane w całym genomie z rozdzielczością 100’s lub 1000’s bp. Istnieją również problemy związane z tendencyjnością amplifikacji fragmentów DNA ChIP, normalizacją danych macierzy i porównywalnością platform macierzy.
Jak działa ChIP-seq?
Sekwencjonowanie metodą immunoprecypitacji chromatyny, czyli ChIP-seq, łączy ChIP z sekwencjonowaniem następnej generacji (Barski 2007, Johnson 2007). Protokoły ChIP-seq zostały zaadaptowane z metod ChIP-chip: białka są usieciowane do związanego z nimi DNA przez traktowanie formaldehydem, komórki są homogenizowane, a chromatyna jest ścinana i immunoprecypitowana za pomocą kulek magnetycznych związanych z przeciwciałami. Immunoprecypitowane DNA jest następnie wykorzystywane jako dane wejściowe do protokołu przygotowania biblioteki sekwencjonowania następnej generacji, gdzie jest sekwencjonowane i analizowane pod kątem miejsc wiążących DNA. Zobacz poniższy rysunek, aby zapoznać się z podsumowaniem przepływu pracy ChIP-seq i przykład wyników ChIP-seq (reprodukowane za uprzejmą zgodą Dominica Schmidta (Schmidt 2009)).
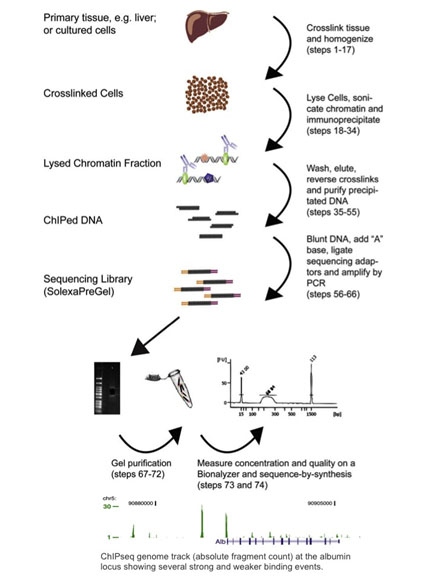
Chociaż większość z około 400 opublikowanych do tej pory prac analizowano na platformie Illumina, ChIP-seq można wykonywać na dowolnym sekwenatorze następnej generacji (Wold 2008). ChIP-seq została szeroko przyjęta od czasu jej pierwszego zgłoszenia w 2007 roku. W rzeczywistości prawie całkowicie wyparła ChIP-Chip, ponieważ umożliwia analizę całego genomu i nie ma ograniczeń omówionych powyżej.
Co właściwie można zrobić z ChIP-seq
ChIP-seq jest potężnym i wszechstronnym narzędziem, a w literaturze można znaleźć wiele wspaniałych przykładów wykorzystania ChIP-seq. Wybrałam kilka moich ulubionych (z pracy wykonanej w ośrodku, którym zarządzam), aby zilustrować to, co jest możliwe, i zawarłam przykłady, w których ChIP-seq zainspirował rozwój nowych metod:
- Antoni Hurtado, et al. wykonali knock-down “czynnika pionierskiego” FoxA1, co spowodowało zmniejszenie wiązania przez receptor estrogenowy (ER) w ponad 50% znanych miejsc wiązania ER. Wykazali, że FoxA1 jest ważnym regulatorem transkrypcji indukowanej przez ER, co sugeruje, że może być nowym i ważnym celem terapeutycznym w raku piersi (Hurtado 2011).
- Dominic Shmidt i wsp. użyli ChIP-seq do zbadania ewolucji wiązania czynników transkrypcyjnych. Skoncentrowali się na wiązaniu CEBPA i HNF4 w tkance wątroby pięciu gatunków kręgowców: człowieka, myszy, psa, oposa i kurczaka. ChIP-chip byłby prawie niemożliwy, biorąc pod uwagę różne zaangażowane gatunki i złożoność w projektowaniu sond (Schmidt 2010).
Dalsze modyfikacje metody ChIP-seq doprowadziły do powstania kilku nowych metod, w tym ChIP-seq do analizy interakcji RNA-białko, a także DNase-seq i FAIRE-seq, z których obie są wykorzystywane do identyfikacji regionów regulatorowych w DNA.
Podsumowując, ChIP-seq jest dojrzałą techniką, która jest lepsza od ChIP-chip i może być używana przez prawie każdą grupę zainteresowaną analizą interakcji DNA:Białko. Jak mógłbyś wykorzystać ChIP-seq w swoich badaniach?
Barski et al, High-resolution profiling of histone methylations in the human genome. Cell 129 (2007).
Hurtado et al, FOXA1 is a key determinant of estrogen receptor function and endocrine response.Nature Genetics (2011).
Johnson et al, Genome-wide mapping of in vivo protein-DNA interactions. Science (2007).
Lee et al, Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc (2006).
Lieb et al, Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001.
Morozova & Marra, Applications of next-generation sequencing technologies in functional genomics. Genomics (2008).
Schmidt et al, ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods (2009).
Schmidt et al, Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding Science (2010).
Wold & Myers, Sequence census methods for functional genomics. Nat. Methods (2008).
Czy to Ci pomogło? Następnie proszę podzielić się z siecią.
.